Role of FBXW7 in the quiescence of gefitinib-resistant lung cancer stem cells in EGFR-mutant non-small cell lung cancer
DOI:
https://doi.org/10.17305/bjbms.2019.4227Keywords:
FBXW7, quiescence, cancer stem cells, gefitinib resistance, NSCLCAbstract
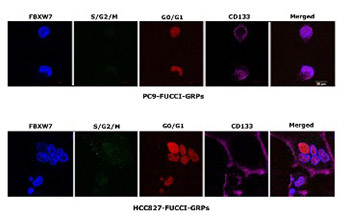
Several recent studies suggest that cancer stem cells (CSCs) are involved in intrinsic resistance to cancer treatment. Maintenance of quiescence is crucial for establishing resistance of CSCs to cancer therapeutics. F-box/WD repeat-containing protein 7 (FBXW7) is a ubiquitin ligase that regulates quiescence by targeting the c-MYC protein for ubiquitination. We previously reported that gefitinib-resistant persisters (GRPs) in EGFR-mutant non-small cell lung cancer (NSCLC) cells highly expressed octamer-binding transcription factor 4 (Oct-4) as well as the lung CSC marker CD133, and they exhibited distinctive features of the CSC phenotype. However, the role of FBXW7 in lung CSCs and their resistance to epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors in NSCLC is not fully understood. In this study, we developed GRPs from the two NSCLC cell lines PC9 and HCC827, which express an EGFR exon 19 deletion mutation, by treatment with a high concentration of gefitinib. The GRPs from both PC9 and HCC827 cells expressed high levels of CD133 and FBXW7, but low levels of c-MYC. Cell cycle analysis demonstrated that the majority of GRPs existed in the G0/G1 phase. Knockdown of the FBXW7 gene significantly reduced the cell number of CD133-positive GRPs and reversed the cell population in the G0/G1-phase. We also found that FBXW7 expression in CD133-positive cells was increased and c-MYC expression was decreased in gefitinib-resistant tumors of PC9 cells in mice and in 9 out of 14 tumor specimens from EGFR-mutant NSCLC patients with acquired resistance to gefitinib. These findings suggest that FBXW7 plays a pivotal role in the maintenance of quiescence in gefitinib-resistant lung CSCs in EGFR mutation-positive NSCLC.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
How to Cite
Accepted 2019-05-18
Published 2019-11-08





