Pathogenesis of atherosclerosis in the tunica intima, media, and adventitia of coronary arteries: An updated review
DOI:
https://doi.org/10.17305/bjbms.2019.4320Keywords:
Atherosclerosis, intima, media, adventitia, perivascular adipose tissueAbstract
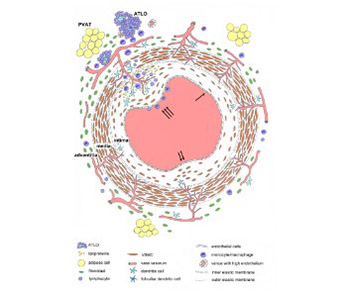
Atherosclerosis is a chronic inflammatory disease of arteries and it affects the structure and function of all three layers of the coronary artery wall. Current theories suggest that the dysfunction of endothelial cells is one of the initial steps in the development of atherosclerosis. The view that the tunica intima normally consists of a single layer of endothelial cells attached to the subendothelial layer and internal elastic membrane has been questioned in recent years. The structure of intima changes with age and it becomes multilayered due to migration of smooth muscle cells from the media to intima. At this stage, the migration and proliferation of smooth muscle cells do not cause pathological changes in the intima. The multilayering of intima is classically considered to be an important stage in the development of atherosclerosis, but in fact atherosclerotic plaques develop only focally due to the interplay of various processes that involve the resident and invading inflammatory cells. The tunica media consists of multiple layers of smooth muscle cells that produce the extracellular matrix, and this layer normally does not contain microvessels. During the development of atherosclerosis, the microvessels from the tunica adventitia or from the lumen may penetrate thickened media to provide nutrition and oxygenation. According to some theories, the endothelial dysfunction of these nutritive vessels may significantly contribute to the atherosclerosis of coronary arteries. The adventitia contains fibroblasts, progenitor cells, immune cells, microvessels, and adrenergic nerves. The degree of inflammatory cell infiltration into the adventitia, which can lead to the formation of tertiary lymphoid organs, correlates with the severity of atherosclerotic plaques. Coronary arteries are surrounded by perivascular adipose tissue that also participates in the atherosclerotic process.
Citations
Downloads
Downloads
Additional Files
Published
How to Cite
Accepted 2019-07-31
Published 2020-02-05





