Apoptosis in pancreatic β-islet cells in Type 2 diabetes
DOI:
https://doi.org/10.17305/bjbms.2016.919Keywords:
Amyloid, apoptosis, β-cells, caspase, hyperglycemia, insulin, islets of Langerhans, knockout mouse, pro-apoptotic genes, Type 2 diabetesAbstract
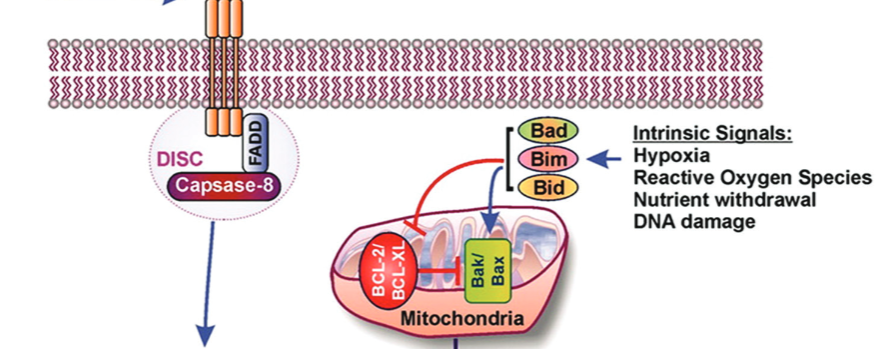
Apoptosis plays important roles in the pathophysiology of Type 2 diabetes mellitus (T2DM). The etiology of T2DM is multifactorial, including obesity-associated insulin resistance, defective insulin secretion, and loss of β-cell mass through β-cell apoptosis. β-cell apoptosis is mediated through a milliard of caspase family cascade machinery in T2DM. The glucose-induced insulin secretion is the principle pathophysiology of diabetes and insufficient insulin secretion results in chronic hyperglycemia, diabetes. Recently, hyperglycemia-induced β-cell apoptosis has been extensively studied on the balance of pro-apoptotic Bcl-2 proteins (Bad, Bid, Bik, and Bax) and anti-apoptotic Bcl family (Bcl-2 and Bcl-xL) toward apoptosis in vitro isolated islets and insulinoma cell culture. Apoptosis can only occur when the concentration of pro-apoptotic Bcl-2 exceeds that of anti-apoptotic proteins at the mitochondrial membrane of the intrinsic pathway. A bulk of recent research on hyperglycemia-induced apoptosis on β-cells unveiled complex details on glucose toxicity on β-cells in molecular levels coupled with cell membrane potential by adenosine triphosphate generation through K+ channel closure, opening Ca2+ channel and plasma membrane depolarization. Furthermore, animal models using knockout mice will shed light on the basic understanding of the pathophysiology of diabetes as a glucose metabolic disease complex, on the balance of anti-apoptotic Bcl family and pro-apoptotic genes. The cumulative knowledge will provide a better understanding of glucose metabolism at a molecular level and will lead to eventual prevention and therapeutic application for T2DM with improving medications.
Citations
Downloads
References
Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26(4):239-57.
http://dx.doi.org/10.1038/bjc.1972.33.
Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas: Estimation with a simple mathematical model. Diabetes 1995; 44(3):249-56.
http://dx.doi.org/10.2337/diab.44.3.249.
Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 1997; 138(4):1736-41.
http://dx.doi.org/10.1210/endo.138.4.5069.
Mandrup-Poulsen T. beta-cell apoptosis: Stimuli and signaling. Diabetes 2001; 50 Suppl 1:S58-63. http://dx.doi.org/10.2337/diabetes.50.2007.S58.
Lee SC, Pervaiz S. Apoptosis in the pathophysiology of diabetes mellitus. Int J Biochem Cell Biol 2007; 39(3):497-504. http://dx.doi.org/10.1016/j.biocel.2006.09.007.
Emamaullee JA, Shapiro AM. Interventional strategies to prevent beta-cell apoptosis in islet transplantation. Diabetes 2006; 55(7):1907-14. http://dx.doi.org/10.2337/db05-1254.
Nakano M, Matsumoto I, Sawada T, Ansite J, Oberbroeckling J, Zhang HJ, et al. Caspase-3 inhibitor prevents apoptosis of human islets immediately after isolation and improves islet graft function. Pancreas 2004; 29(2):104-9. http://dx.doi.org/10.1097/00006676-200408000-00004.
Brandhorst D, Kumarasamy V, Maatoui A, Alt A, Bretzel RG, Brandhorst H. Porcine islet graft function is affected by pretreatment with a caspase-3 inhibitor. Cell Transplant 2006; 15(4):311-7. http://dx.doi.org/10.3727/000000006783981936.
Cheng G, Zhu L, Mahato RI. Caspase-3 gene silencing for inhibiting apoptosis in insulinoma cells and human islets. Mol Pharm 2008; 5(6):1093-102. http://dx.doi.org/10.1021/mp800093f.
Nicholson DW, Thornberry NA. Caspases: Killer proteases. Trends Biochem Sci 1997; 22(8):299-306. http://dx.doi.org/10.1016/S0968-0004(97)01085-2.
Chandra J, Zhivotovsky B, Zaitsev S, Juntti-Berggren L, Berggren PO, Orrenius S. Role of apoptosis in pancreatic beta-cell death in diabetes. Diabetes 2001; 50 Suppl 1:S44-7.
http://dx.doi.org/10.2337/diabetes.50.2007.S44.
Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol 1998; 10(5):545-51. http://dx.doi.org/10.1016/S0952-7915(98)80222-7.
Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998; 391(6662):96-9. http://dx.doi.org/10.1038/34214.
Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev 1999; 13(15):1899-911. http://dx.doi.org/10.1101/gad.13.15.1899.
Green DR. Apoptotic pathways: Ten minutes to dead. Cell 2005; 121(5):671-4.
http://dx.doi.org/10.1016/j.cell.2005.05.019.
Federici M, Hribal M, Perego L, Ranalli M, Caradonna Z, Perego C, et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: A potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes 2001; 50(6):1290-301. http://dx.doi.org/10.2337/diabetes.50.6.1290.
Wajant H. The Fas signaling pathway: More than a paradigm. Science 2002; 296(5573):1635-6. http://dx.doi.org/10.1126/science.1071553.
Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell 1993; 75(6):1169-78.
http://dx.doi.org/10.1016/0092-8674(93)90326-L.
Allison J, Thomas HE, Catterall T, Kay TW, Strasser A. Transgenic expression of dominant-negative Fas-associated death domain protein in beta cells protects against Fas ligand- induced apoptosis and reduces spontaneous diabetes in nonobese diabetic mice. J Immunol 2005; 175(1):293-301. http://dx.doi.org/10.4049/jimmunol.175.1.293.
Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev 2008; 29(3):303-16. http://dx.doi.org/10.1210/er.2007-0037.
Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988; 37(12):1595-607. http://dx.doi.org/10.2337/diab.37.12.1595.
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52(1):102-10. http://dx.doi.org/10.2337/diabetes.52.1.102.
Bonner-Weir S. Islet growth and development in the adult. J Mol Endocrinol 2000; 24(3):297-302.
http://dx.doi.org/10.1677/jme.0.0240297.
Marchetti P, Del Guerra S, Marselli L, Lupi R, Masini M, Pollera M, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab 2004; 89(11):5535-41.
http://dx.doi.org/10.1210/jc.2004-0150.
Tomita T, Iwata K. Gelatinases and inhibitors of gelatinases in pancreatic islets and islet cell tumors. Modern Pathol 1997; 10(1):47-54.
Perez SE, Cano DA, Dao-Pick T, Rougier JP, Werb Z, Hebrok M. Matrix metalloproteinases 2 and 9 are dispensable for pancreatic islet formation and function in vivo. Diabetes 2005; 54(3):694-701. http://dx.doi.org/10.2337/diabetes.54.3.694.
Hirata H, Takahashi A, Kobayashi S, Yonehara S, Sawai H, Okazaki T, et al. Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis. J Exp Med 1998; 187(4):587-600. http://dx.doi.org/10.1084/jem.187.4.587.
Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 1995; 81(5):801-9.
http://dx.doi.org/10.1016/0092-8674(95)90541-3.
Gown AM, Willingham MC. Improved detection of apoptotic cells in archival paraffin sections: Immunohistochemistry using antibodies to cleaved caspase 3. J Histochem Cytochem 2002;50(4):449-54. http://dx.doi.org/10.1177/002215540205000401.
Martin SJ, Green DR. Protease activation during apoptosis: Death by a thousand cuts? Cell 1995; 82(3):349-52. http://dx.doi.org/10.1016/0092-8674(95)90422-0.
Tomita T. Immunocytochemical localisation of caspase-3 in pancreatic islets from type 2 diabetic subjects. Pathology 2010; 42(5):432-7.
http://dx.doi.org/10.3109/00313025.2010.493863.
Bonner-Weir S. Life and death of the pancreatic beta cells. Trends Endocrinol Metab 2000; 11(9):375-8. http://dx.doi.org/10.1016/S1043-2760(00)00305-2.
Bonner-Weir S, O'Brien TD. Islets in type 2 diabetes: In honor of Dr. Robert C. Turner. Diabetes 2008; 57(11):2899-904. http://dx.doi.org/10.2337/db07-1842.
Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death? Science 2005; 307(5708):380-4. http://dx.doi.org/10.1126/science.1104345.
Tomita T. Immunocytochemical localization of cleaved caspase-3 in pancreatic islets from type 1 diabetic subjects. Islets 2010; 2(1):24-9. http://dx.doi.org/10.4161/isl.2.1.10041.
Labat-Moleur F, Guillermet C, Lorimier P, Robert C, Lantuejoul S, Brambilla E, et al. TUNEL apoptotic cell detection in tissue sections: Critical evaluation and improvement. J Histochem Cytochem 1998; 46(3):327-34. http://dx.doi.org/10.1177/002215549804600306.
Duan WR, Garner DS, Williams SD, Funckes-Shippy CL, Spath IS, Blomme EA. Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J Pathol 2003; 199(2):221-8. http://dx.doi.org/10.1002/path.1289.
Tomita T. Cleaved caspase-3 immunocytochemical staining for pancreatic islets and pancreatic endocrine tumors: A potential marker for biological malignancy. Islets 2010; 2(2):82-8. http://dx.doi.org/10.4161/isl.2.2.10807.
Ehrlich JC, Ratner IM. Amyloidosis of the islets of Langerhans. A restudy of islet hyalin in diabetic and non-diabetic individuals. Am J Pathol 1961; 38(1):49-59.
Höppener JW, Ahrén B, Lips CJ. Islet amyloid and type 2 diabetes mellitus. N Engl J Med 2000; 343(9):411-9.
Hull RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: A critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab 2004; 89(8):3629-43.
http://dx.doi.org/10.1210/jc.2004-0405.
Butler AE, Janson J, Soeller WC, Butler PC. Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: Evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 2003; 52(9):2304-14.
http://dx.doi.org/10.2337/diabetes.52.9.2304.
Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A 1987; 84(23):8628-32. http://dx.doi.org/10.1073/pnas.84.23.8628.
Westermark P, Wernstedt C, Wilander E, Sletten K. A novel peptide in the calcitonin gene related peptide family as an amyloid fibril protein in the endocrine pancreas. Biochem Biophys Res Commun 1986; 140(3):827-31. http://dx.doi.org/10.1016/0006-291X(86)90708-4.
Kahn SE, Andrikopoulos S, Verchere CB. Islet amyloid: A long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 1999; 48(2):241-53.
http://dx.doi.org/10.2337/diabetes.48.2.241.
Buse JB, Weyer C, Maggs DC. Amylin replacement with Pramlintide in type 1 and type 2 diabetes: A physiological approach to overcome a barrier with insulin therapy. Clin Diabetes 2002; 20(3):137-44. http://dx.doi.org/10.2337/diaclin.20.3.137.
Fineman M, Weyer C, Maggs DG, Strobel S, Kolterman OG. The human amylin analog, pramlintide, reduces postprandial hyperglucagonemia in patients with type 2 diabetes mellitus. Horm Metab Res 2002; 34(9):504-8. http://dx.doi.org/10.1055/s-2002-34790.
Kruger DF, Gatcomb PM, Owen SK. Clinical implications of amylin and amylin deficiency. Diabetes Educ 1999; 25(3):389-97. http://dx.doi.org/10.1177/014572179902500310.
Weyer C, Maggs DG, Young AA, Kolterman OG. Amylin replacement with pramlintide as an adjunct to insulin therapy in type 1 and type 2 diabetes mellitus: A physiological approach toward improved metabolic control. Curr Pharm Des 2001; 7(14):1353-73.
http://dx.doi.org/10.2174/1381612013397357.
Tomita T. Islet amyloid polypeptide in pancreatic islets from type 2 diabetic subjects. Islets 2012; 4(3):223-32. http://dx.doi.org/10.4161/isl.20477.
Clark A, Nilsson MR. Islet amyloid: A complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia 2004; 47(2):157-69.
http://dx.doi.org/10.1007/s00125-003-1304-4.
Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis quantitative changes in the pancreas in type 2 diabetes. Diabetes Res 1988; 9(4):151-9.
Jurgens CA, Toukatly MN, Fligner CL, Udayasankar J, Subramanian SL, Zraika S, et al. ß-cell loss and ß-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol 2011; 178(6):2632-40. http://dx.doi.org/10.1016/j.ajpath.2011.02.036.
Mirzabekov TA, Lin MC, Kagan BL. Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem 1996; 271(4):1988-92. http://dx.doi.org/10.1074/jbc.271.4.1988.
O’Brien TD, Glabe CG, Butler PC. Evidence for proteotoxicity in beta cells in type 2 diabetes: Toxic islet amyloid oligomers from intracellularly in the secretory pathway. Am J Pathol 2010; 176(2):861-9.
Anguiano M, Nowak RJ, Lansbury PT Jr. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry 2002; 41(38):11338-43. http://dx.doi.org/10.1021/bi020314u.
Engel MF, Khemtémourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, et al. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci U S A 2008; 105(16):6033-8. http://dx.doi.org/10.1073/pnas.0708354105.
Jansen A, van Hagen M, Drexhage HA. Defective maturation and function of antigen-presenting cells in type 1 diabetes. Lancet 1995; 345(8948):491-2.
http://dx.doi.org/10.1016/S0140-6736(95)90586-3.
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300(5618):486-9. http://dx.doi.org/10.1126/science.1079469.
Ritzel RA, Butler PC. Replication increases beta-cell vulnerability to human islet amyloid polypeptide-induced apoptosis. Diabetes 2003; 52:1701-8.
http://dx.doi.org/10.2337/diabetes.52.7.1701.
Tomita T. Amylin in human adult pancreatic islets. Pathology 2003; 35(1):34-6.
http://dx.doi.org/10.1097/01268031-200335010-00005, http://dx.doi.org/10.1080/0031302021000062299.
Tomita T, Lacy PE, Matschinsky FM, McDaniel ML. Effect of alloxan on insulin secretion in isolated rat islets perifused in vitro. Diabetes 1974; 23(6):517-24.
http://dx.doi.org/10.2337/diab.23.6.517.
Tomita T, Scarpelli DG. Interaction of cyclic AMP and alloxan on insulin secretion in isolated rat islets perifused in vitro. Endocrinology 1977; 100(5):1327-33.
http://dx.doi.org/10.1210/endo-100-5-1327.
Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005; 54 Suppl 2:S97-107.
http://dx.doi.org/10.2337/diabetes.54.suppl_2.S97.
Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, et al. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005; 54 Suppl 2:S108-13.
http://dx.doi.org/10.2337/diabetes.54.suppl_2.S108.
Bedoya FJ, Matschinsky FM, Shimizu T, O'Neil JJ, Appel MC. Differential regulation of glucokinase activity in pancreatic islets and liver of the rat. J Biol Chem 1986; 261(23):10760-4.
Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes 1990; 39(6):647-52.
http://dx.doi.org/10.2337/diab.39.6.647,
http://dx.doi.org/10.2337/diabetes.39.6.647.
Matschinsky F, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, et al. Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J Clin Invest 1993; 92(5):2092-8.
http://dx.doi.org/10.1172/JCI116809.
Shimizu T, Knowles BB, Matschinsky FM. Control of glucose phosphorylation and glucose usage in clonal insulinoma cells. Diabetes 1988; 37(5):563-8.
http://dx.doi.org/10.2337/diab.37.5.563.
MacDonald PE, Joseph JW, Rorsman P. Glucose-sensing mechanisms in pancreatic beta-cells. Philos Trans R Soc Lond B Biol Sci 2005; 360(1464):2211-25.
http://dx.doi.org/10.1098/rstb.2005.1762.
De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, et al. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest 1995; 96(5):2489-95. http://dx.doi.org/10.1172/JCI118308.
Iynedjian PB. Mammalian glucokinase and its gene. Biochem J 1993; 293:1-13.
http://dx.doi.org/10.1042/bj2930001.
Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem 1995; 64:689-719.
http://dx.doi.org/10.1146/annurev.bi.64.070195.003353.
Fridlyand L, Philson LH. Glucose sensing in the pancreatic beta cells: A computational systems analysis. Theor Biol Med Model 2010; 7(15):1-44.
http://dx.doi.org/10.1186/1742-4682-7-15.
Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature 1992; 356(6365):162-4. http://dx.doi.org/10.1038/356162a0.
Porter JR, Barrett TG. Monogenic syndromes of abnormal glucose homeostasis: Clinical review and relevance to the understanding of the pathology of insulin resistance and beta cell failure. J Med Genet 2005; 42(12):893-902.
http://dx.doi.org/10.1136/jmg.2005.030791.
Tattersall R. Maturity-onset diabetes of the young: A clinical history. Diabet Med 1998; 15(1):11-4.
http://dx.doi.org/10.1002/(SICI)1096-9136(199801)15:1<11::AID-DIA561>3.0.CO;2-0.
What is Maturity-Onset Diabetes of the Young (MODY)? National Diabetes Information Clearinghouse (NDIC) (National Institute of Diabetes and Digestive and Kidney Diseases) (NIH). [Last retrieved on 2008 Jul 29].
Leahy JL, Bonner-Weir S, Weir GC. Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care 1992; 15(3):442-55. http://dx.doi.org/10.2337/diacare.15.3.442.
Kim WH, Lee JW, Suh YH, Hong SH, Choi JS, Lim JH, et al. Exposure to chronic high glucose induces beta-cell apoptosis through decreased interaction of glucokinase with mitochondria: Downregulation of glucokinase in pancreatic beta-cells. Diabetes 2005; 54(9):2602-11.
http://dx.doi.org/10.2337/diabetes.54.9.2602.
Chan SL, Yu VC. Proteins of the bcl-2 family in apoptosis signalling: From mechanistic insights to therapeutic opportunities. Clin Exp Pharmacol Physiol 2004; 31(3):119-28.
http://dx.doi.org/10.1111/j.1440-1681.2004.03975.x.
Hengartner MO. The biochemistry of apoptosis. Nature 2000; 407(6805):770-6.
http://dx.doi.org/10.1038/35037710.
Ou D, Wang X, Metzger DL, James RF, Pozzilli P, Plesner A, et al. Synergetic inhibition of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human pancreatic beta cells by Bcl-2 and X-linked inhibitor of apoptosis. Hum Immunol 2005; 66(3):274-84.
http://dx.doi.org/10.1016/j.humimm.2004.12.002.
Saldeen J. Cytokines induce both necrosis and apoptosis via a common Bcl-2-inhibitable pathway in rat insulin-producing cells. Endocrinology 2000; 141(6):2003-10.
http://dx.doi.org/10.1210/endo.141.6.7523.
Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol 2003; 15(6):725-31. http://dx.doi.org/10.1016/j.ceb.2003.10.009.
Hui H, Dotta F, Di Mario U, Perfetti R. Role of caspases in the regulation of apoptotic pancreatic islet beta-cells death. J Cell Physiol 2004; 200(2):177-200.
http://dx.doi.org/10.1002/jcp.20021.
Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003; 424(6951):952-6. http://dx.doi.org/10.1038/nature01825.
Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJ, et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med 2008; 14(2):144-53. http://dx.doi.org/10.1038/nm1717.
Antinozzi PA, Ishihara H, Newgard CB, Wollheim CB. Mitochondrial metabolism sets the maximal limit of fuel-stimulated insulin secretion in a model pancreatic beta cell: A survey of four fuel secretagogues. J Biol Chem 2002; 277(14):11746-55.
http://dx.doi.org/10.1074/jbc.M108462200.
Berggren PO, Larsson O. Ca2+ and pancreatic B-cell function. Biochem Soc Trans 1994; 22(1):12-8.
http://dx.doi.org/10.1042/bst0220012.
Liang Y, Bai N, Doliba C, Wang L, Barner DK, Matschinsky FM. Glucose metabolism and insulin release in mouse HC9 cells, as model of wild-type pancreatic beta cells. Am J Physiol 1996; 270:E846-57.
Luciani DS, White SA, Widenmaier SB, Saran VV, Taghizadeh F, Hu X, et al. Bcl-2 and Bcl-xL suppress glucose signaling in pancreatic ß-cells. Diabetes 2013; 62(1):170-82.
http://dx.doi.org/10.2337/db11-1464.
Real PJ, Cao Y, Wang R, Nikolovska-Coleska Z, Sanz-Ortiz J, Wang S, et al. Breast cancer cells can evade apoptosis-mediated selective killing by a novel small molecule inhibitor of Bcl-2. Cancer Res 2004; 64(21):7947-53.
http://dx.doi.org/10.1158/0008-5472.CAN-04-0945.
Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, et al. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell 2011; 146(4):607-20.
http://dx.doi.org/10.1016/j.cell.2011.06.050.
Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A 2009; 106(34):14397-402.
http://dx.doi.org/10.1073/pnas.0907555106.
Simonson DC. Hyperinsulinemia and its sequelae. Horm Metab Res Suppl 1990; 22:17-25.
Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 2008; 29(1):42-61. http://dx.doi.org/10.1210/er.2007-0015.
Thomas HE, McKenzie MD, Angstetra E, Campbell PD, Kay TW. Beta cell apoptosis in diabetes. Apoptosis 2009; 14(12):1389-404. http://dx.doi.org/10.1007/s10495-009-0339-5.
Zhou YP, Pena JC, Roe MW, Mittal A, Levesetti M, Baldwin AC, et al. Overexpression of Bcl-xL in beta cells prevents cell death but impairs mitochondrial signal for insulin secretion. Am J Physiol Endocrinol Metab 2000; 278(2):E340-51.
Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005; 307(5708)384-7.
http://dx.doi.org/10.1126/science.1104343.
Dunn CJ, Peters DH. Metformin. A review of its pharmacological properties and therapeutic use in non-insulin-dependent diabetes mellitus. Drugs 1995; 49(5):721-49.
Hundal RS, Inzucchi SE. Metformin: New understandings, new uses. Drugs 2003; 63(18):1879-94.
http://dx.doi.org/10.2165/00003495-200363180-00001.
Gunton JE, Delhanty PJ, Takahashi SI, Baxter RC. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2 and increase glucose uptake via increased GLUT-2 translocation. J Clin Endocrinol Metab 2003; 88(3):323-32.
http://dx.doi.org/10.1210/jc.2002-021394.
Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes 2002; 51 Suppl 3:S368-76.
http://dx.doi.org/10.2337/diabetes.51.2007.S368.
Kendall DM. Thiazolidinediones: The case for early use. Diabetes Care 2006; 29(1):154-7. http://dx.doi.org/10.2337/diacare.29.01.06.dc05-0711.
Ahrén B, Schmitz O. GLP-1 receptor agonists and DPP-4 inhibitors in the treatment of type 2 diabetes. Horm Metab Res 2004; 36(11-12):867-76. http://dx.doi.org/10.1055/s-2004-826178.
Abdul-Ghani MA, Norton L, DeFrenzo RA. Role of sodium-glucose cotransporter 2 (SGLT-2) inhibitors in the treatment of type 2 diabetes. Endocr Rev 2011; 32(4):515-31.
Downloads
Additional Files
Published
License
Copyright (c) 2016 Bosnian Journal of Basic Medical Sciences

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2016-01-20
Published 2016-08-02





