Potential inflammatory markers in obstructive sleep apnea-hypopnea syndrome
DOI:
https://doi.org/10.17305/bjbms.2016.1579Keywords:
Obstructive sleep apnea-hypopnea syndrome, hypoxia-inducible factor-1, nuclear factor-κB, surfactant proteinAbstract
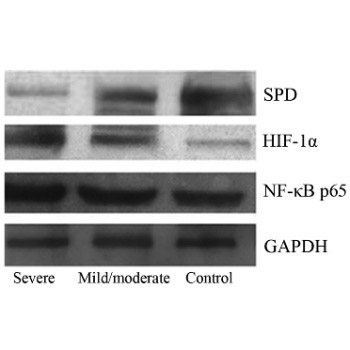
Obstructive sleep apnea-hypopnea syndrome (OSAHS) is a complex chronic inflammatory respiratory disease with multiple pathogenic factors and high morbidity and mortality. Serum levels of nuclear factor-κB (NF-κB), hypoxia-inducible factor-1 alpha (HIF-1α), and surfactant protein D (SPD) were investigated in OSAHS patients, to determine their clinical significance and correlation with the pathogenesis. Patients were classified into a mild and moderate OSAHS group (n = 25) and severe OSAHS group (n = 33). Twenty healthy patients served as a control group. Peripheral blood levels of NF-κB, HIF-1α, and SPD were determined by Western blot, and a correlation analysis was performed. Severe OSAHS patients received nasal continuous positive airway pressure (nCPAP) therapy and were followed up after 2 months. NF-κB p65, HIF-1α, and SPD expression levels were determined after valid nCPAP therapy. NF-κB p65 and HIF-1α expression was significantly higher in severe OSAHS group than in the other two groups (p < 0.01), and was positively correlated with the apnea-hypopnea index (AHI) (r = 0.696, p < 0.001; r = 0.634, p < 0.001). SPD expression was significantly lower in severe OSAHS group than in the control group (p < 0.01) and mild and moderate OSAHS group (p < 0.01), and was negatively correlated with AHI (r = −0.569, p < 0.001). OSAHS pathogenesis was associated with changes in NF-κB, HIF-1α, and SPD protein expression levels. nCPAP therapy could improve the clinical characteristics of the patients, lower serum NF-κB and HIF-1α levels, and increase serum SPD levels. We conclude that OSAHS is related to the expression of NF-κB, HIF-1, and SPD.
Citations
Downloads
References
Daulatzai MA. Role of sensory stimulation in amelioration of obstructive sleep apnea. Sleep Disord 2011;2011:596879. http://dx.doi.org/10.1155/2011/596879.
Katz ES, D'Ambrosio CM. Pathophysiology of pediatric obstructive sleep apnea. Proc Am Thorac Soc 2008;5(2):253-62. http://dx.doi.org/10.1513/pats.200707-111MG.
Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc 2008;5(2):136-43. http://dx.doi.org/10.1513/pats.200709-155MG.
Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013;177(9):1006-14. http://dx.doi.org/10.1093/aje/kws342.
Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 2005;112(17):2660-7. http://dx.doi.org/10.1161/CIRCULATIONAHA.105.556746.
Winnicki M, Shamsuzzaman A, Lanfranchi P, Accurso V, Olson E, Davison D, et al. Erythropoietin and obstructive sleep apnea. Am J Hypertens 2004;17(9):783-6. http://dx.doi.org/10.1016/j.amjhyper.2004.04.011.
van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J 2008;412(3):477-84. http://dx.doi.org/10.1042/BJ20080476.
Belaiba RS, Bonello S, Zähringer C, Schmidt S, Hess J, Kietzmann T, et al. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell 2007;18(12):4691-7.
http://dx.doi.org/10.1091/mbc.E07-04-0391.
Bonello S, Zähringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, et al. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol 2007;27(4):755-61. http://dx.doi.org/10.1161/01.ATV.0000258979.92828.bc.
Arnardottir ES, Mackiewicz M, Gislason T, Teff KL, Pack AI. Molecular signatures of obstructive sleep apnea in adults: A review and perspective. Sleep 2009;32(4):447-70.
Cochrane CG, Spragg R, Revak SD. Pathogenesis of the adult respiratory distress syndrome. Evidence of oxidant activity in bronchoalveolar lavage fluid. J Clin Invest 1983;71(3):754-761. http://dx.doi.org/10.1172/JCI110823.
Kiefmann R, Rifkind JM, Nagababu E, Bhattacharya J. Red blood cells induce hypoxic lung inflammation. Blood 2008;111(10):5205-14. http://dx.doi.org/10.1182/blood-2007-09-113902.
Ito Y, Ahmad A, Kewley E, Mason RJ. Hypoxia-inducible factor regulates expression of surfactant protein in alveolar type II cells in vitro. Am J Respir Cell Mol Biol 2011;45(5):938-45. http://dx.doi.org/10.1165/rcmb.2011-0052OC.
Ghadiali SN, Gaver DP. Biomechanics of liquid-epithelium interactions in pulmonary airways. Respir Physiol Neurobiol 2008;163(1-3):232-43. DOI: 10.1016/j.resp.2008.04.008.
Tenner AJ, Robinson SL, Borchelt J, Wright JR. Human pulmonary surfactant protein (SP-A), a protein structurally homologous to Clq, can enhance FcR- and CR1-mediated phagocytosis. J Biol Chem 1989;264(23):13923-8.
van Iwaarden F, Welmers B, Verhoef J, Haagsman HP, van Golde LM. Pulmonary surfactant protein A enhances the host-defense mechanism of rat alveolar macrophages. Am J Respir Cell Mol Biol 1990;2(1):91-8. http://dx.doi.org/10.1165/ajrcmb/2.1.91.
Hohlfeld J, Fabel H, Hamm H. The role of pulmonary surfactant in obstructive airways disease. Eur Respir J 1997;10(2):482-91. http://dx.doi.org/10.1183/09031936.97.10020482.
Antal JM, Divis LT, Erzurum SC, Wiedemann HP, Thomassen MJ. Surfactant suppresses NF-kappa B activation in human monocytic cells. Am J Respir Cell Mol Biol 1996;14(4):374-9. http://dx.doi.org/10.1165/ajrcmb.14.4.8600942.
Phelps DS, Floros J. Localization of pulmonary surfactant proteins using immunohistochemistry and tissue in situ hybridization. Exp Lung Res 1991;17(6):985-95. http://dx.doi.org/10.3109/01902149109064330.
Takahashi H, Fujishima T, Koba H, Murakami S, Kurokawa K, Shibuya Y, et al. Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med 2000;162(3 Pt 1):1109-14.
Stapleton RD, Dixon AE, Parsons PE, Ware LB, Suratt BT; NHLBI Acute Respiratory Distress Syndrome Network. The association between BMI and plasma cytokine levels in patients with acute lung injury. Chest 2010;138(3):568-77. http://dx.doi.org/10.1378/chest.10-0014.
Winkler C, Atochina-Vasserman EN, Holz O, Beers MF, Erpenbeck VJ, Krug N, et al. Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir Res 2011;12:29. DOI: 10.1186/1465-9921-12-29.
He QY, Chen BY. Sleep related breathing disorders team, respiology branch, Chinese medical association, OSAHS diagnosis and treatment guideline (revision 2011) [Article in Chinese]. Chinese J Tuberc Respir Dis 2012;35(1):9-12.
Ciccone MM, Scicchitano P, Mitacchione G, Zito A, Gesualdo M, Caputo P, et al. Is there a correlation between OSAS duration/severity and carotid intima-media thickness? Respir Med 2012;106(5):740-6. http://dx.doi.org/10.1016/j.rmed.2011.12.016.
Atkeson A, Jelic S. Mechanisms of endothelial dysfunction in obstructive sleep apnea. Vasc Health Risk Manag 2008;4(6):1327-35.
https://dx.doi.org/10.2147/VHRM.S4078.
Damiani MF, Carratù P, Quaranta VN, Resta O. Influence of obstructive sleep apnea on endothelial function in obese patients. Chest 2012;141:1639; author reply 1640. http://dx.doi.org/10.1378/chest.12-0144.
Sarada SKS, Veeramohan PH, Mathew T, Saumya S, Chitharanjan M. Nifedipine inhibits hypoxia induced transvascular leakage through down regulation of NFkB. Respir Physiol Neurobiol 2012;183(1):26-34. http://dx.doi.org/10.1016/j.resp.2012.05.016.
Fitzpatrick SF, Tambuwala MM, Bruning U, Schaible B, Scholz CC, Byrne A, et al. An intact canonical NF-kB pathway is required for inflammatory gene expression in response to hypoxia. J Immunol 2011;186(2):1091-6. http://dx.doi.org/10.4049/jimmunol.1002256.
Htoo AK, Greenberg H, Tongia S, Chen G, Henderson T, Wilson D, et al. Activation of nuclear factor kappaB in obstructive sleep apnea: A pathway leading to systemic inflammation. Sleep Breath 2006;10(1):43-50.
http://dx.doi.org/10.1007/s11325-005-0046-6.
Ciccone MM, Scicchitano P, Zito A, Cortese F, Boninfante B, Falcone VA, et al. Correlation between inflammatory markers of atherosclerosis and carotid intima-media thickness in obstructive sleep apnea. Molecules 2014;19(2):1651-62. http://dx.doi.org/10.3390/molecules19021651.
Li RC, Haribabu B, Mathis SP, Kim J, Gozal D. Leukotriene B4 receptor-1 mediates intermittent hypoxia-induced atherogenesis. Am J Respir Crit Care Med 2011;184(1):124-31. http://dx.doi.org/10.1164/rccm.201012-2039OC.
Dyugovskaya L, Polyakov A, Ginsberg D, Lavie P, Lavie L. Molecular pathways of spontaneous and TNF-{alpha}-mediated neutrophil apoptosis under intermittent hypoxia. Am J Respir Cell Mol Biol 2011;45(1):154-62. http://dx.doi.org/10.1165/rcmb.2010-0025OC.
Greenberg H, Ye X, Wilson D, Htoo AK, Hendersen T, Liu SF. Chronic intermittent hypoxia activates nuclear factor-kappaB in cardiovascular tissues in vivo. Biochem Biophys Res Commun 2006;343(2):591-6. http://dx.doi.org/10.1016/j.bbrc.2006.03.015.
Carratù P, Zito A, Dragonieri S, Ciccone MM, Resta O. Effect of CPAP on cardiovascular risks in OSA patients. A 4-year follow-up, preliminary data. Sleep Breath 2016. [Epub ahead of print]. http://dx.doi.org/10.1007/s11325-016-1382-4.
Sin DD, Pahlavan PS, Man SF. Surfactant protein D: A lung specific biomarker in COPD? Ther Adv Respir Dis 2008;2(2):65-74. http://dx.doi.org/10.1177/1753465808088903.
Wirtz HR, Dobbs LG. Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science 1990;250(4985):1266-9. http://dx.doi.org/10.1126/science.2173861.
Wirtz H, Schmidt M. Ventilation and secretion of pulmonary surfactant. Clin Investig 1992;70(1):3-13. http://dx.doi.org/10.1007/BF00422930.
Massaro GD, Massaro D. Morphologic evidence that large inflations of the lung stimulate secretion of surfactant. Am Rev Respir Dis 1983;127(2):235-6.
Aderibigbe AO, Thomas RF, Mercer RR, Auten RL Jr. Brief exposure to 95% oxygen alters surfactant protein D and mRNA in adult rat alveolar and bronchiolar epithelium. Am J Respir Cell Mol Biol 1999;20(2):219-27. http://dx.doi.org/10.1165/ajrcmb.20.2.3348.
Liang S, Li N, Heizhati M, Yao X, Abdireim A, Wang Y, et al. What do changes in concentrations of serum surfactant proteins A and D in OSA mean? Sleep Breath 2015;19(3):955-62. http://dx.doi.org/10.1007/s11325-014-1106-6.
Downloads
Additional Files
Published
Issue
Section
Categories
How to Cite
Accepted 2016-09-13
Published 2017-02-21





