Defective autophagy is associated with neuronal injury in a mouse model of multiple sclerosis
DOI:
https://doi.org/10.17305/bjbms.2017.1696Keywords:
Autophagy, experimental autoimmune encephalomyelitis, multiple sclerosis, neurodegeneration, apoptosis, EAE mice, LC3-II, Beclin1, rapamycin, 3-methyladenine, p62, autophagy deficiencyAbstract
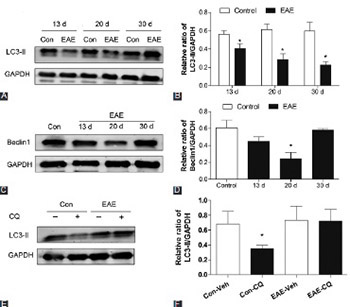
Neurodegeneration, along with inflammatory demyelination, is an important component of multiple sclerosis (MS) pathogenesis. Autophagy is known to play a pivotal role in neuronal homeostasis and is implicated in several neurodegenerative disorders. However, whether autophagy is involved in the mechanisms of neuronal damage during MS remains to be investigated. Experimental autoimmune encephalomyelitis (EAE), an in vivo model of MS, was induced in female C57BL/6 mice by immunization with myelin oligodendrocyte glycoprotein p35-55. After that, autophagic flux in the spinal cord of mice was evaluated by detection of LC3-II and Beclin1 protein expressions. EAE mice were then administered with rapamycin and 3-methyladenine (3-MA) for 10 days. Afterward, the changes in LC3-II, Beclin1, and p62 expression, number of infiltrated inflammatory cells, demyelinated lesion area, and neuronal damage, as well as clinical scores, were assessed. Further, apoptotic cell rate and apoptosis-related protein expressions were monitored. We observed an impaired autophagic flux and increased neuronal damage in the spinal cords of EAE mice. We also found that rapamycin, an autophagy inducer, mitigated EAE-induced autophagy decrease, inflammation, demyelination and neuronal injury, as well as the abnormal clinical score. In addition, rapamycin suppressed cell apoptosis, and decreased Bax/Bcl-2 ratio and cleaved caspase-3 expression. Conversely, the effect of autophagy inhibitor 3-MA on EAE mice resulted in completely opposite results. These results indicated that autophagy deficiency, at least in part, contributed to EAE-induced neuronal injury and that pharmacological modulation of autophagy might be a therapeutic strategy for MS.
Citations
Downloads
References
Obert D, Helms G, Sättler MB, Jung K, Kretzschmar B, Bähr M, et al. Brain metabolite changes in patients with relapsing-remitting and secondary progressive multiple sclerosis: A two-year follow-up study. PLoS One 2016;11(9):e0162583. https://doi.org/10.1371/journal.pone.0162583.
Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: Results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996;46(4):907-11. https://doi.org/10.1212/WNL.46.4.907.
Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011;469(7330):323-35. https://doi.org/10.1038/nature09782.
Bhattacharya A, Eissa NT. Autophagy and autoimmunity crosstalks. Front Immunol 2013;4:88. https://doi.org/10.3389/fimmu.2013.00088.
Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010;13(7):805-11. https://doi.org/10.1038/nn.2575.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441(7095):880-4. https://doi.org/10.1038/nature04723.
Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol 2007;6(4):352-61.
https://doi.org/10.1016/S1474-4422(07)70076-5.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006;441(7095):885-9. https://doi.org/10.1038/nature04724.
Li Z, Chen L, Niu X, Liu J, Ping M, Li R, et al. Immunomodulatory synergy by combining atorvastatin and rapamycin in the treatment of experimental autoimmune encephalomyelitis (EAE). J Neuroimmunol 2012;250(1-2):9-17. https://doi.org/10.1016/j.jneuroim.2012.05.008.
Zhen C, Feng X, Li Z, Wang Y, Li B, Li L, et al. Suppression of murine experimental autoimmune encephalomyelitis development by 1,25-dihydroxyvitamin D3 with autophagy modulation. J Neuroimmunol 2015;280:1-7. https://doi.org/10.1016/j.jneuroim.2015.01.012.
Zhang X, Chen S, Song L, Tang Y, Shen Y, Jia L, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014;10(4):588-602. https://doi.org/10.4161/auto.27710.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000;19(21):5720-8. https://doi.org/10.1093/emboj/19.21.5720.
Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007;26(10):2527-39. https://doi.org/10.1038/sj.emboj.7601689.
Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005;5(9):726-34. https://doi.org/10.1038/nrc1692.
Solomon VR, Lee H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol 2009;625(1-3):220-33. https://doi.org/10.1016/j.ejphar.2009.06.063.
Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005;171(4):603-14. https://doi.org/10.1083/jcb.200507002.
Donia M, Mangano K, Amoroso A, Mazzarino MC, Imbesi R, Castrogiovanni P, et al. Treatment with rapamycin ameliorates clinical and histological signs of protracted relapsing experimental allergic encephalomyelitis in Dark Agouti rats and induces expansion of peripheral CD4 CD25 Foxp3 regulatory T cells. J Autoimmun 2009;33(2):135-40. https://doi.org/10.1016/j.jaut.2009.06.003.
Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: Mechanisms and functional consequences. Curr Opin Neurol 2001;14(3):271-8. https://doi.org/10.1097/00019052-200106000-00003.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007;8(9):741-52. https://doi.org/10.1038/nrm2239.
Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ 2012;19(1):144-52. https://doi.org/10.1038/cdd.2011.78.
Boland B, Nixon RA. Neuronal macroautophagy: From development to degeneration. Mol Aspects Med 2006;27(5-6):503-19. https://doi.org/10.1016/j.mam.2006.08.009.
Akhtar RS, Ness JM, Roth KA. Bcl-2 family regulation of neuronal development and neurodegeneration. Biochim Biophys Acta 2004;1644(2-3):189-203. https://doi.org/10.1016/j.bbamcr.2003.10.013.
Walensky LD. BCL-2 in the crosshairs: Tipping the balance of life and death. Cell Death Differ 2006;13(8):1339-50. https://doi.org/10.1038/sj.cdd.4401992.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005;122(6):927-39. https://doi.org/10.1016/j.cell.2005.07.002.
Raisova M, Hossini AM, Eberle J, Riebeling C, Wieder T, Sturm I, et al. The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J Invest Dermatol 2001;117(2):333-40.
https://doi.org/10.1046/j.0022-202x.2001.01409.x.
Das A, Guyton MK, Matzelle DD, Ray SK, Banik NL. Time-dependent increases in protease activities for neuronal apoptosis in spinal cords of Lewis rats during development of acute experimental autoimmune encephalomyelitis. J Neurosci Res 2008;86(13):2992-3001. https://doi.org/10.1002/jnr.21737.
Kingham PJ, Cuzner ML, Pocock JM. Apoptotic pathways mobilized in microglia and neurones as a consequence of chromogranin A-induced microglial activation. J Neurochem 1999;73(2):538-47. https://doi.org/10.1046/j.1471-4159.1999.0730538.x.
Guyton MK, Das A, Samantaray S, Wallace GC 4th, Butler JT, Ray SK, et al. Calpeptin attenuated inflammation, cell death, and axonal damage in animal model of multiple sclerosis. J Neurosci Res 2010;88(11):2398-408. https://doi.org/10.1002/jnr.22408.
Ahmed Z, Doward AI, Pryce G, Taylor DL, Pocock JM, Leonard JP, et al. A role for caspase-1 and -3 in the pathology of experimental allergic encephalomyelitis: Inflammation versus degeneration. Am J Pathol 2002;161(5):1577-86. https://doi.org/10.1016/S0002-9440(10)64436-7.
Luessi F, Siffrin V, Zipp F. Neurodegeneration in multiple sclerosis: Novel treatment strategies. Expert Rev Neurother 2012;12(9):1061-76. https://doi.org/10.1586/ern.12.59.
Downloads
Additional Files
Published
How to Cite
Accepted 2016-11-15
Published 2017-05-20





