Apoptosis of pancreatic β-cells in Type 1 diabetes
DOI:
https://doi.org/10.17305/bjbms.2017.1961Keywords:
Apoptosis, autoimmunity, β-cells, Bcl family, caspases, cytokines, insulitis, Type 1 diabetesAbstract
Type 1 diabetes mellitus (T1DM) results from autoimmune destruction of pancreatic β-cells after an asymptomatic period over years. Insulitis activates antigen presenting cells, which trigger activating CD4+ helper-T cells, releasing chemokines/cytokines. Cytokines activate CD8+ cytotoxic–T cells, which lead to β-cell destruction. Apoptosis pathway consists of extrinsic (receptor-mediated) and intrinsic (mitochondria-driven) pathway. Extrinsic pathway includes Fas pathway to CD4+-CD8+ interaction, whereas intrinsic pathway includes mitochondria-driven pathway at a balance between anti-apoptotic B-cell lymphoma (Bcl)-2 and Bcl-xL and pro-apoptotic Bad, Bid, and Bik proteins. Activated cleaved caspse-3 is the converging point between extrinsic and intrinsic pathway. Apoptosis takes place only when pro-apoptotic proteins exceed anti-apoptotic proteins. Since the concordance rate of T1DM in identical twins is about 50%, environmental factors are involved in the development of T1DM, opening a door to find means to detect and prevent further development of autoimmune β-cell destruction for a therapeutic application.
Citations
Downloads
References
Daneman D. Type 1 diabetes. Lancet 2006;367(9513):847-58. https://doi.org/10.1016/S0140-6736(06)68341-4.
Yoon JW, Jun HS. Autoimmune destruction of pancreatic beta cells. Am J Ther 2005;12(6):580-91. https://doi.org/10.1097/01.mjt.0000178767.67857.63.
Pirot P, Cardozo AK, Eizirik DL. Mediators and mechanisms of pancreatic beta-cell death in Type 1 diabetes. Arq Bras Endocrinol Metabol 2008;52(2):156-65. https://doi.org/10.1590/S0004-27302008000200003.
Jahromi MM, Eisenbarth GS. Cellular and molecular pathogenesis of Type 1A diabetes. Cell Mol Life Sci 2007;64(7-8):865-72. https://doi.org/10.1007/s00018-007-6469-4.
Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in Type 1 and Type 2 diabetes: Many differences, few similarities. Diabetes 2005;54(Suppl 2):S97-107. https://doi.org/10.2337/diabetes.54.suppl_2.S97.
Kim MS, Polychronakos C. Immunogenetics of Type 1 diabetes. Horm Res 2005;64(4):180-8. https://doi.org/10.1159/000089190.
Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 2007;39(7):857-64. https://doi.org/10.1038/ng2068.
Turner D. The human leucocyte antigen (HLA) system. Vox Sang 2004;87(Suppl 1):87-90. https://doi.org/10.1111/j.1741-6892.2004.00438.x.
Pihoker C, Gilliam LK, Hampe CS, Lernmark A. Autoantibodies in diabetes. Diabetes 2005;54(Suppl 2):S52-61. https://doi.org/10.2337/diabetes.54.suppl_2.S52.
Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human Type 1 diabetes. Proc Natl Acad Sci U S A 2007;104(43):17040-5. https://doi.org/10.1073/pnas.0705894104.
Filippi CM, von Herrath MG. Viral trigger for type 1 diabetes: Pros and cons. Diabetes 2008;57(11):2863-71. https://doi.org/10.2337/db07-1023.
In't Veld P, Lievens D, De Grijse J, Ling Z, Van der Auwera B, Pipeleers-Marichal M, et al. Screening for insulitis in adult autoantibody-positive organ donors. Diabetes 2007;56(9):2400-4. https://doi.org/10.2337/db07-0416.
Knip M, Veijola R, Virtanen SM, Hyöty H, Vaarala O, Akerblom HK. Environmental triggers and determinants of Type 1 diabetes. Diabetes 2005;54(Suppl 2):S125-36. https://doi.org/10.2337/diabetes.54.suppl_2.S125.
Lee SC, Pervaiz S. Apoptosis in the pathophysiology of diabetes mellitus. Int J Biochem Cell Biol 2007;39(3):497-504. https://doi.org/10.1016/j.biocel.2006.09.007.
Nakano M, Matsumoto I, Sawada T, Ansite J, Oberbroeckling J, Zhang HJ, et al. Caspase-3 inhibitor prevents apoptosis of human islets immediately after isolation and improves islet graft function. Pancreas 2004;29(2):104-9.
https://doi.org/10.1097/00006676-200408000-00004.
Cheng G, Zhu L, Mahato RI. Caspase-3 gene silencing for inhibiting apoptosis in insulinoma cells and human islets. Mol Pharm 2008;5(6):1093-102. https://doi.org/10.1021/mp800093f.
Nicholson DW, Thornberry NA. Caspases: Killer proteases. Trends Biochem Sci 1997;22(8):299-306. https://doi.org/10.1016/S0968-0004(97)01085-2.
Chandra J, Zhivotovsky B, Zaitsev S, Juntti-Berggren L, Berggren PO, Orrenius S. Role of apoptosis in pancreatic beta-cell death in diabetes. Diabetes 2001;50(Suppl 1):S44-7. https://doi.org/10.2337/diabetes.50.2007.S44.
Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol 1998;10(5):545-51. https://doi.org/10.1016/S0952-7915(98)80222-7.
Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev 1999;13:1899-911. https://doi.org/10.1101/gad.13.15.1899.
Green DR. Apoptotic pathways: Ten minutes to dead. Cell 2005;121(5):671-4. https://doi.org/10.1016/j.cell.2005.05.019.
Federici M, Hribal M, Perego L, Ranalli M, Caradonna Z, Perego C, et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: A potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes 2001;50(6):1290-301. https://doi.org/10.2337/diabetes.50.6.1290.
Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, et al. Human ICE/CED-3 protease nomenclature. Cell 1996;87(2):171. https://doi.org/10.1016/S0092-8674(00)81334-3.
Stennicke HR, Salvesen GS. Biochemical characteristics of caspases-3, -6, -7, and -8. J Biol Chem 1997;272(41):25719-23. https://doi.org/10.1074/jbc.272.41.25719.
Katunuma N, Matsui A, Le QT, Utsumi K, Salvesen G, Ohashi A. Novel procaspase-3 activating cascade mediated by lysoapoptases and its biological significances in apoptosis. Adv Enzyme Regul 2001;41:237-50. https://doi.org/10.1016/S0065-2571(00)00018-2.
Li P, Nijhawn D, Wang X. Mitochondrial activation of apoptosis. Cell 2004;116(Suppl 21):s57-9. https://doi.org/10.1016/S0092-8674(04)00031-5.
Hirata H, Takahashi A, Kobayashi S, Yonehara S, Sawai H, Okazaki T, et al. Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis. J Exp Med 1998;187(4):587-600. https://doi.org/10.1084/jem.187.4.587.
Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 1995;81(5):801-9.
https://doi.org/10.1016/0092-8674(95)90541-3.
Cell Signaling Technology. Cleaved Caspase-3 (Asp 175) Antibody, Cat No. 9661. Vol. 9661. Beverly, MA: Cell Signaling Technology; 2006.
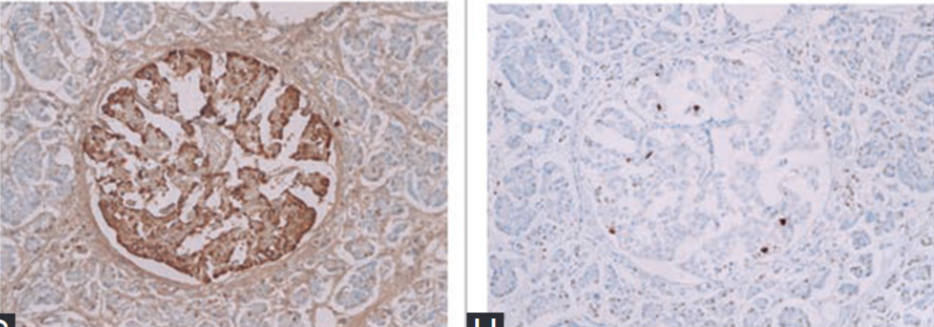
Tomita T. Caspase-3 immunocytochemical staining for pancreatic islets and pancreatic endocrine tumors. Hum Pathol 2009;40(7):1050-2. https://doi.org/10.1016/j.humpath.2009.02.010.
Gown AM, Willingham MC. Improved detection of apoptotic cells in archival paraffin sections: Immunohistochemistry using antibodies to cleaved caspase3. J Histochem Cytochem 2002;50(4):449-54. https://doi.org/10.1177/002215540205000401.
Martin SJ, Green DR. Protease activation during apoptosis: Death by a thousand cuts? Cell 1995;82(3):349-52. https://doi.org/10.1016/0092-8674(95)90422-0.
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with Type 2 diabetes. Diabetes 2003;52(1):102-10. https://doi.org/10.2337/diabetes.52.1.102.
Tomita T. Immunocytochemical localization of cleaved caspase-3 in pancreatic islets from Type 1 diabetic subjects. Islets 2010;2(1):24-9. https://doi.org/10.4161/isl.2.1.10041.
Tomita T. Immunocytochemical localisation of caspase-3 in pancreatic islets from Type 2 diabetic subjects. Pathology 2010;42(5):432-7. https://doi.org/10.3109/00313025.2010.493863.
Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing Type 1 diabetes: Indirect evidence for islet regeneration? Diabetologia 2005;48(11):2221-8. https://doi.org/10.1007/s00125-005-1949-2.
Butler AE, Galasso R, Meier JJ, Basu R, Rizza RA, Butler PC. Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset Type 1 diabetic patients who died of diabetic ketoacidosis. Diabetologia 2007;50(11):2323-31. https://doi.org/10.1007/s00125-007-0794-x.
Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death? Science 2005;307 (5708):380-4. https://doi.org/10.1126/science.1104345.
Adams JM, Cory S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998;281(5381):1322-6. https://doi.org/10.1126/science.281.5381.1322.
Zamzami N, Brenner C, Marzo I, Susin SA, Kroemer G. Subcellular and submitochondrial mode of action of Bcl-2-like oncoproteins. Oncogene 1998;16(17):2265-82. https://doi.org/10.1038/sj.onc.1201989.
Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta 2004;1644(2-3):83-94. https://doi.org/10.1016/j.bbamcr.2003.08.012.
Kim WH, Lee JW, Suh YH, Hong SH, Choi JS, Lim JH, et al. Exposure to chronic high glucose induces beta-cell apoptosis through decreased interaction of glucokinase with mitochondria: Downregulation of glucokinase in pancreatic beta-cells. Diabetes 2005;54(9):2602-11. https://doi.org/10.2337/diabetes.54.9.2602.
Chan SL, Yu VC. Proteins of the bcl-2 family in apoptosis signalling: From mechanistic insights to therapeutic opportunities. Clin Exp Pharmacol Physiol 2004;31(3):119-28. https://doi.org/10.1111/j.1440-1681.2004.03975.x.
Hengartner MO. The biochemistry of apoptosis. Nature 2000;407(6805):770-6. https://doi.org/10.1038/35037710.
Ou D, Wang X, Metzger DL, James RF, Pozzilli P, Plesner A, et al. Synergetic inhibition of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human pancreatic beta cells by Bcl-2 and X-linked inhibitor of apoptosis. Hum Immunol 2005;66(3):274-84. https://doi.org/10.1016/j.humimm.2004.12.002.
Saldeen J. Cytokines induce both necrosis and apoptosis via a common Bcl-2-inhibitable pathway in rat insulin-producing cells. Endocrinology 2000;141(6):2003-10. https://doi.org/10.1210/en.141.6.2003; https://doi.org/10.1210/endo.141.6.7523.
Hui H, Dotta F, Di Mario U, Perfetti R. Role of caspases in the regulation of apoptotic pancreatic islet beta-cells death. J Cell Physiol 2004;200(2):177-200. https://doi.org/10.1002/jcp.20021.
Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003;424(6951):952-6. https://doi.org/10.1038/nature01825.
Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJ, et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med 2008;14(2):144-53. https://doi.org/10.1038/nm1717.
Araki E, Oyadomari S, Mori M. Endoplasmic reticulum stress and diabetes mellitus. Intern Med 2003;42(1):7-14. https://doi.org/10.2169/internalmedicine.42.7.
Downloads
Additional Files
Published
License
Copyright (c) 2017 Bosnian Journal of Basic Medical Sciences

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2017-02-19
Published 2017-08-20





