Differential diagnosis of hepatopulmonary syndrome (HPS): Portopulmonary hypertension (PPH) and hereditary hemorrhagic telangiectasia (HHT)
DOI:
https://doi.org/10.17305/bjbms.2017.2020Keywords:
Hepatopulmonary syndrome, differential diagnosis, portopulmonary hypertension, hereditary hemorrhagic telangiectasia, PPH, HPS, HHT, orthotopic liver transplantation, OLTAbstract
Hepatopulmonary syndrome (HPS) is a severe complication of advanced liver disease associated with an extremely poor prognosis. HPS is diagnosed in 4-47% of patients with cirrhosis and in 15-20% of candidates for liver transplantation. In addition, severe hypoxia is associated with a high risk of complications of liver transplantation (a 30% chance during the first 90 days) and increases the gap between transplantation and improving arterial oxygenation. The pathogenesis of HPS is not fully understood, and no effective pharmacological treatment has been developed yet. Currently, the treatment of choice for HPS is orthotopic liver transplantation. Non-specific clinical criteria and the lack of standardized diagnostic criteria for determining HPS can lead to diagnostic errors. Portopulmonary hypertension and hereditary hemorrhagic telangiectasia, also known as Osler–Weber–Rendu syndrome, are pulmonary complications of liver disease which should be differentially diagnosed from HPS.
Citations
Downloads
References
Fluckiger M. Vorkommen von trommelschagel formigen fingerendphalangen ohne chronische veranderungen an der lungen oder am herzen. Wien Med Wochenschr. 1884;34:1457.
Snell AM. The effects of chronic disease of the liver on the composition and physiochemical properties of blood: Changes in the serum proteins; reduction in the oxygen saturation of the arterial blood. Ann Intern Med 1935;9(6):690-711. https://doi.org/10.7326/0003-4819-9-6-690.
Rydell R, Hoffbauer FW. Multiple pulmonary arteriovenous fistulas in juvenile cirrhosis. Am J Med 1956;21(3):450-60. https://doi.org/10.1016/0002-9343(56)90043-2.
Kennedy TC, Knudson RJ. Exercise-aggravated hypoxemia and orthodeoxia in cirrhosis. Chest 1977;72(3):305-9. https://doi.org/10.1378/chest.72.3.305.
Grace JA, Angus PW. Hepatopulmonary syndrome: Update on recent advances in pathophysiology, investigation, and treatment. J Gastroenterol Hepatol 2013;28(2):213-9. https://doi.org/10.1111/jgh.12061.
Martınez-Palli G, Rodrıguez-Roisin R. Hepatopulmonary syndrome: A liver induced oxygenation defect. Eur Respir Mon 2011;54:246-64. https://doi.org/10.1183/1025448x.10008510.
Rodriguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome – A liver-induced lung vascular disorder. N Engl J Med 2008;358(22):2378-87. https://doi.org/10.1056/NEJMra0707185.
Zhang J, Fallon MB. Hepatopulmonary syndrome: Update on pathogenesis and clinical features. Nat Rev Gastroenterol Hepatol 2012;9(9):539-49. https://doi.org/10.1038/nrgastro.2012.123.
Ilchenko LY, Fedorov IG, Karabinenko AA. Gepatopulmonalnyy sindrom: Sostoyaniye problemy [Hepatopulmonary syndrome: State of problem]. Sovremennyye tekhnologii v meditsine 2009;1:84-8.
Dinh-Xuan AT, Naeije R. The hepatopulmonary syndrome: No way out? Eur Respir J 2004;23(5):661-2. https://doi.org/10.1183/09031936.04.00028204.
Rodriguez-Roisin R, Krowka MJ, Herve PH, Fallon MB, ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD). Eur Respir J 2004;24(5):861-80. https://doi.org/10.1183/09031936.04.00010904.
Lima B, Martinelli A, Franca AV. Hepatopulmonary syndrome: Pathogenesis, diagnosis and treatment [Article in Portuguese]. Arq Gastroenterol 2004;41(4):250-8. https://doi.org/10.1590/S0004-28032004000400010.
Ivashkin VT, Morozova MA, Mayevskaya MV. Gepatopulmonalnyy sindrom [Hepatopulmonary syndrome]. Transplantologiya 2009;2:5-8.
Shafiq M, Khan AA, Alam A, Butt AK, Shafqat F, Malik K, et al. Frequency of hepatopulmonary syndrome in cirrhotic patients. J Coll Physicians Surg Pak 2008;18(5):278-81. DOI: 05.2008/JCPSP.278281.
Macêdo LG, Lopes EP. Hepatopulmonary syndrome: An update. Sao Paulo Med J 2009;127(4):223-30. https://doi.org/10.1590/S1516-31802009000400008.
Roberts KE, Kawut SM, Krowka MJ, Brown RS, Trotter JF, Shah V, et al. Genetic risk factors for hepatopulmonary syndrome in patients with advanced liver disease. Gastroenterology 2010;139(1):130-9. https://doi.org/10.1053/j.gastro.2010.03.044.
Krowka MJ, Cortese DA. Hepatopulmonary syndrome: Current concepts in diagnostic and therapeutic considerations. Chest 1994;105(5):1528-37. https://doi.org/10.1378/chest.105.5.1528.
Lange PA, Stoller JK. The hepatopulmonary syndrome. Ann Intern Med 1995;122(7):521-9. https://doi.org/10.7326/0003-4819-122-7-199504010-00008.
Schenk P, Schöniger-Hekele M, Fuhrmann V, Madl C, Silberhumer G, Müller C. Prognostic significance of the hepatopulmonary syndrome in patients with cirrhosis. Gastroenterology 2003;125(4):1042-52. https://doi.org/10.1016/S0016-5085(03)01207-1.
Teuber G, Teupe C, Dietrich CF, Caspary WF, Buhl R, Zeuzem S. Pulmonary dysfunction in non-cirrhotic patients with chronic viral hepatitis. Eur J Intern Med 2002;13(5):311-8. https://doi.org/10.1016/S0953-6205(02)00066-3.
Shulpekova YO, Sokolina IA. Gepatopulmonalnyy sindrom: Patologicheskaya fiziologiya, klinicheskoye techeniye, diagnostika i lecheniye [Hepatopulmonary syndrome: Pathological physiology, clinical course, diagnostics and treatment]. Klinicheskiye Perspektivy Gastroenterologii, Gepatologii 2006;4:16-21.
De BK, Sen S, Biswas PK, Mandal SK, Das D, Das U, et al. Occurrence of hepatopulmonary syndrome in Budd-Chiari syndrome and the role of venous decompression. Gastroenterology 2002;122(4):897-903. https://doi.org/10.1053/gast.2002.32419.
Regev A, Yeshurun M, Rodriguez M, Sagie A, Neff GW, Molina EG, et al. Transient hepatopulmonary syndrome in a patient with acute hepatitis A. J Viral Hep 2001;8(1):83-6.
Umeda N, Kamath PS. Hepatopulmonary syndrome and portopulmonary hypertension. Hepatol Res 2009;39(10):1020-2. https://doi.org/10.1111/j.1872-034X.2009.00552.x.
Babbs C, Warnes TW, Haboubu NY. Non-cirrhotic portal hypertension with hypoxaemia. Gut 1988;29(1):129-31. https://doi.org/10.1136/gut.29.1.129.
Sari S, Oguz D, Sucak T, Dalgic B, Atasever T. Hepatopulmonary syndrome in children with cirrhotic and non-cirrhotic portal hypertension: A single-center experience. Dig Dis Sci 2012;57(1):175-81. https://doi.org/10.1007/s10620-011-1832-6.
Herve P, Lebrec D, Brenot F, Simonneau G, Humbert M, Sitbon O, et al. Pulmonary vascular disorders in portal hypertension. Europ Respir J 1998;11(5):1153-66. https://doi.org/10.1183/09031936.98.11051153.
Lau VWS, Lau DCY, Huen KF. Hepatopulmonary syndrome: An unusual presentation of chronic hypervitaminosis A. J Paediatr 2008;13:46-52.
Avendano CE, Flume PA, Baliga P, Lewin DN, Strange C, Reuben A. Hepatopulmonary syndrome occurring after orthotopic liver transplantation. Liver Transpl 2001;7(12):1081-4. https://doi.org/10.1053/jlts.2001.29416.
Patil V, Cherian G. Hepatopulmonary syndrome, severe cyanosis and marfanoid habitus. J Assoc Physicians India 2014;62(12):57-60.
Lange PA, Stoller JK. The hepatopulmonary syndrome. Ann Intern Med 1995;122(7):521-9. https://doi.org/10.7326/0003-4819-122-7-199504010-00008.
Alizadeh AHM, Fatemi SR, Mirzaee V, Khoshbaten M, Talebipour B, Sharifian A, et al. Clinical features of hepatopulmonary syndrome in cirrhotic patients. World J Gastroenterol 2006;12(12):1954-6. https://doi.org/10.3748/wjg.v12.i12.1954.
Harris EA, Kenyon AM, Nisbet HD, Seelye ER, Whitlock RM. The normal alveolar-arterial oxygen-tension gradient in man. Clin Sci Mol Med 1974;46(1):89-104. https://doi.org/10.1042/cs0460089.
Zhang ZJ, Yang CQ. Progress in investigating the pathogenesis of hepatopulmonary syndrome. Hepatobiliary Pancreat Dis Int 2010;9(4):355-60.
Zamirian M, Aslani A, Shahrzad S. Left atrial volume: A novel predictor of hepatopulmonary syndrome. Am J Gastroenterol 2007;102(7):1392-6. https://doi.org/10.1111/j.1572-0241.2007.01228.x.
Pouriki S, Alexopoulou A, Chrysochoou C, Raftopoulos L, Papatheodoridis G, Stefanadis C, et al. Left ventricle enlargement and increased systolic velocity in the mitral valve are indirect markers of the hepatopulmonary syndrome. Liver Int 2011;31(9):1388-94. https://doi.org/10.1111/j.1478-3231.2011.02591.x.
Dziedziczko A, Bartuzi Z. The hepatopulmonary syndrome - known symptoms and new name. Case Rep Clin Pract Rev 2002;3(2):121-7.
Arguedas MR, Singh H, Faulk DK, Fallon MB. Utility of pulse oximetry screening for hepatopulmonary syndrome. Clin Gastroenterol Hepatol 2007;5(6):749-54. https://doi.org/10.1016/j.cgh.2006.12.003.
Deibert P, Allgaier HP, Loesch S, Müller C, Olschewski M, Hamm H, et al. Hepatopulmonary syndrome in patients with chronic liver disease: Role of pulse oximetry. BMC Gastroenterol 2006;6:15. https://doi.org/10.1186/1471-230X-6-15.
Horvatits T, Drolz A, Roedl K, Herkner H, Ferlitsch A, Perkmann T, et al. Von Willebrand factor antigen for detection of hepatopulmonary syndrome in patients with cirrhosis. J Hepatol 2014;61(3):544-9. https://doi.org/10.1016/j.jhep.2014.04.025.
Horvatits T, Drolz A, Rutter K, Roedl K, Fauler G, Müller C, et al. Serum bile acids in patients with hepatopulmonary syndrome. Z Gastroenterol 2017;55(4):361-7. DOI: 10.1055/s-0042-121268.
Ho V. Current concepts in the management of hepatopulmonary syndrome. Vasc Health Risk Manag 2008;4(5):1035-41. https://doi.org/10.2147/VHRM.S3608.
American Thoracic Society Executive Committee. Recommended standardized procedures for pulmonary testing. Amer Rev Respir Dis 1978;118:55-72.
Abragamovich MO. Gepatopulmonalniy sindrom: osoblivostí patog̀enezu, díag̀nostiki, klíníchnogo perebígu ta líkuvannya [Hepatopulmonalnyy syndrome: features of patogenesis, diagnostics, clinical course and treatment]. Ukrayinskyy medychnyy almanakh 2010;13(5):10-3.
Kamath PS. Portopulmonary hypertension and hepatopulmonary syndrome. J Gastroenterol Hepatol 2002;17(3):S253-5. https://doi.org/10.1046/j.1440-1746.17.s3.9.x.
Fuhrmann V, Drolz A, Rutter K, Horvatits T. HPS: Diagnosis, clinical features, and medical therapy. Clin Liver Dis 2014;4(2):46-9. https://doi.org/10.1002/cld.402.
Espinosa MD, Nogueras F, Olmedo C, Macias R, Muffak-Granero K, Comino A, et al. Hepatopulmonary syndrome among cirrhotic candidates for liver transplantation. Transplant Proc 2012;44(6):1508-9. https://doi.org/10.1016/j.transproceed.2012.06.001.
Krowka MJ. Management of pulmonary complications in pretransplant patients. Clin Liver Dis 2011;15(4):765-77. https://doi.org/10.1016/j.cld.2011.08.012.
Machicao VI, Fallon MB. Hepatopulmonary syndrome. Semin Respir Crit Care Med 2012;33(1):11-6. https://doi.org/10.1055/s-0032-1301730.
Taillé С, Cadranel J, Bellocq A, Thabut G, Soubrane O, Durand F, et al. Liver transplantation for hepatopulmonary syndrome: A ten-year experience in Paris, France. Transplantation 2003;75(9):1482-9. https://doi.org/10.1097/01.TP.0000061612.78954.6C.
Nayyar D, Man HS, Granton J, Lilly LB, Gupta S. Proposed management algorithm for severe hypoxemia after liver transplantation in the hepatopulmonary syndrome. Am J Transplant 2015;15(4):903-13. https://doi.org/10.1111/ajt.13177.
Fukushima KY, Yatsuhashi H, Kinoshita A, Ueki T, Matsumoto T, Osumi M, et al. Two cases of hepatopulmonary syndrome with improved liver function following long-term oxygen therapy. J Gastroenterol 2007;42(2):176-80.
https://doi.org/10.1007/s00535-006-1965-0.
Arguedas MR, Abrams GA, Krowka MJ, Fallon MB. Prospective evaluation of outcomes and predictors of mortality in patients with hepatopulmonary syndrome undergoing liver transplantation. Hepatology 2003;37(1):192-7. https://doi.org/10.1053/jhep.2003.50023.
Kursov SV, Mykhnevych KH, Lyzohub VN, Skoroplet SN. Gepatopulmonalniy sindrom [Hepatopulmonalnyy syndrome]. Meditsina Neotlozhnykh Sostoyaniy 2009;5(24):35-9.
Swanson KL, Wiesner RH, Krowka MJ. Natural history of hepatopulmonary syndrome: Impact of liver transplantation. Hepatology 2005;41(5):1122-9. https://doi.org/10.1002/hep.20658.
Gupta S, Castel H, Rao RV, Picard M, Lilly L, Faughnan ME, et al. Improved survival after liver transplantation in patients with hepatopulmonary syndrome. Am J Transplant 2010;10(2):354-63. https://doi.org/10.1111/j.1600-6143.2009.02822.x.
Garbuzenko DV. Portopulmonalnaya gipertenziya i gepatopulmonalnyy sindrom u bolnykh tsirrozom pecheni [Portopulmonary hypertension and hepatopulmonary syndrome in patients with cirrhosis]. Pulmonologiya 2006;1:103-7.
Naeije R. Hepatopulmonary syndrome and portopulmonary hypertension. Swiss Med Wkly 2003;133(11-12):163-9.
Colle I, Van Steenkiste C, Geerts A, Van Vlierberghe H. Hepatopulmonary syndrome and portopulmonary hypertension: What's new? Acta Gastroenterol Belg 2007;70(2):203-9.
Abragamovich MO, Abragamovich OO. Porto Pul'monal'na gípertenzíya: osoblivostí patog̀enezu, díag̀nostiki, klíníchnogo perebígu ta líkuvannya [Portopulmonary hypertension: features of patogenesis, diagnostics, clinical course and treatment]. Ukrayinskyy Medychnyy Almanakh 2010;13(4):15-9.
Budhiraja R, Hassoun PM. Portopulmonary hypertension: A tale of two circulations. Chest 2003;123(2):562-76. https://doi.org/10.1378/chest.123.2.562.
Castro M, Krowka MJ, Schroeder DR, Beck KC, Plevak DJ, Rettke SR, et al. Frequency and clinical implications of increased pulmonary artery pressures in liver transplant patients. Mayo Clinic Proc 1996;71(6):543-51. https://doi.org/10.4065/71.6.543.
Mandell MS, Groves BM. Pulmonary hypertension in chronic liver disease. Clin Chest Med 1996;17(1):17-33. https://doi.org/10.1016/S0272-5231(05)70296-3.
Ramsay M. Portopulmonary hypertension and right heart failure in patients with cirrhosis. Curr Opin Anaesthesiol 2010;23(2):145-50. https://doi.org/10.1097/ACO.0b013e32833725c4.
Swanson KL, Wiesner RH, Nyberg SL, Rosen CB, Krowka MJ. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant 2008;8(11):2445-53. https://doi.org/10.1111/j.1600-6143.2008.02384.x.
Ma C, Crippin JS, Chapman WC, Korenblat K, Vachharajani N, Gunter KL, et al. Parenchymal alterations in cirrhotic livers in patients with hepatopulmonary syndrome or portopulmonary hypertension. Liver Transpl 2013;19(7):741-50. https://doi.org/10.1002/lt.23632.
Kawut SM, Krowka MJ, Trotter JF, Roberts KE, Benza RL, Badesch DB, et al. Clinical risk factors for portopulmonary hypertension. Hepatology 2008;48(1):196-203. https://doi.org/10.1002/hep.22275.
Roberts KE, Fallon MB, Krowka MJ, Brown RS, Trotter JF, Peter I, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Amer J Respir Crit Care Med 2009;179(9):835-42. https://doi.org/10.1164/rccm.200809-1472OC.
Møller S, Krag A, Madsen JL, Henriksen JH, Bendtsen F. Pulmonary dysfunction and hepatopulmonary syndrome in cirrhosis and portal hypertension. Liver Int 2009;29(10):1528-37. https://doi.org/10.1111/j.1478-3231.2009.02103.x.
Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004;363(9419):1461-8.
https://doi.org/10.1016/S0140-6736(04)16107-2.
Jamison BM, Michel RP. Different distribution of plexiform lesions in primary and secondary pulmonary hypertension. Hum Pathol 1995;26(9):987-93. https://doi.org/10.1016/0046-8177(95)90088-8.
Edwards BS, Weir EK, Edwards WD, Ludwig J, Dykoski RK, Edwards JE. Coexistent pulmonary and portal hypertension: Morphologic and clinical features. J Amer Coll Cardiol 1987;10(6):1233-8. https://doi.org/10.1016/S0735-1097(87)80123-7.
Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (Gene PPH-1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000;67(3):737-44. https://doi.org/10.1086/303059.
Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic teleangiectasia. N Engl J Med 2001;345(5):325-34. https://doi.org/10.1056/NEJM200108023450503.
Murray KF, Carithers RL Jr, AASLD. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology 2005;41(6):1407-32. https://doi.org/10.1002/hep.20704.
Krowka MJ, Mandell MS, Ramsay MA, Kawut SM, Fallon MB, Manzarbeitia C, et al. Hepatopulmonary syndrome and portopulmonary hypertension: A report of the multicenter liver transplant database. Liver Transpl 2004;10(2):174-82. https://doi.org/10.1002/lt.20016.
Krowka MJ, Plevak DJ, Findlay JY, Rosen CB, Wiesner RH, Krom RA. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl 2000;6(4):443-50. https://doi.org/10.1053/jlts.2000.6356.
Jones FD, Kuo PC, Johnson LB, Njoku MJ, Dixon-Ferguson MK, Plotkin JS. The coexistence of portopulmonary hypertension and hepatopulmonary syndrome. Anesthesiology 1999;90(2):626-9. https://doi.org/10.1097/00000542-199902000-00041.
Shah T, Isaac J, Adams D, Kelly D. Development of hepatopulmonary syndrome and portopulmonary hypertension in a paediatric liver transplantat patient. Pediatr Transplant 2005;9(1):127-31. https://doi.org/10.1111/j.1399-3046.2004.00221.x.
Ioachimescu OC, Mehta AC, Stoller JK. Hepatopulmonary syndrome following portopulmonary hypertension. Eur Respir J 2007;29(6):1277-80. https://doi.org/10.1183/09031936.00140306.
Sharathkumar AA, Shapiro A. Hereditary haemorrhagic telangiectasia. Haemophilia 2008;14(6):1269-80. https://doi.org/10.1111/j.1365-2516.2008.01774.x.
Levandovskyy YA, Tupykyna NV. Nasledstvennaya hemorrahycheskaya teleanhyéktazyya (boleznʹ Randyu-Oslera) [Hereditary hemorrhagic telangiectasia (Rendu-Osler disease)]. Bolezni Serdtsa i Sosudov 2009;3:65-70.
da Silva Santos PS, Fernandes KS, Magalhães MH. Osler-Weber-Rendu syndrome – Dental implications. J Can Dent Assoc 2009;75(7):527-530.
Hanes F. Multiple hereditary telangiectases causing hemorrhage (hereditary hemorrhagic telangiectasia). Bull Johns Hopkins Hosp 1909;20:63-73.
Levandovskyy YA, Antonova MA. Osobennosti klinicheskogo techeniya nasledstvennoy gemorragicheskoy teleangiektazii [Clinical features of hereditary hemorrhagic telangiectasia]. Trudnyy Patsiyent 2007;4:25-8.
Zharkova MS, Lapshin AV, German N. Sosudistyye malformatsii legkikh i pecheni u bolnogo s nasledstvennoy gemorragicheskoy teleangiektaziyey [Vascular malformation of the lungs and liver in a patient with hereditary hemorrhagic telangiectasia]. Ros Zh Gastroenterol Gepatol Koloproktol 2011;2:62-8.
Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000;91(1):66-7. https://doi.org/10.1002/(SICI)1096-8628(20000306)91:1<66::AID-AJMG12>3.0.CO;2-P.
Prigoda NL, Savas S, Abdalla SA, Piovesan B, Rushlow D, Vandezande K, et al. Hereditary haemorrhagic telangiectasia: Mutation detection, test sensitivity and novel mutations. J Med Genet 2006;43(9):722-8. https://doi.org/10.1136/jmg.2006.042606.
Sabbà C, Pasculli G, Lenato GM, Suppressa P, Lastella P, Memeo M, et al. Hereditary hemorrhagic telangiectasia: Clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost 2007;5(6):1149-57. https://doi.org/10.1111/j.1538-7836.2007.02531.x.
Letteboer TGW, Mager HJ, Snijder RJ, Lindhout D, Ploos van Amstel HK, Zanen P, et al. Genotype-phenotype relationship for localization and age distribution of telangiectases in hereditary hemorrhagic telangiectasia. Am J Med Genet 2008;146A(21):2733-9. https://doi.org/10.1002/ajmg.a.32243.
Garcia-Tsao G. Liver involvement in hereditary hemorrhagic teleangiectasia (HHT). J Hepatol 2007;46(3):499-507. https://doi.org/10.1016/j.jhep.2006.12.008.
Manawadu D, Vethanayagam D, Ahmed SN. Hereditary hemorrhagic telangiectasia: Transient ischemic attacks. CMAJ 2009;180(8):836-7. https://doi.org/10.1503/cmaj.081550.
Memeo M, Stabile Ianora AA, Scardapane A, Buonamico P, Sabbà C, Angelelli G. Hepatic involvement in hereditary hemorrhagic telangiectasia: CT findings. Abdom Imaging 2004;29(2):211-20. https://doi.org/10.1007/s00261-003-0101-3.
Levandovskyy YA, Zemskov YV. Sovremennyye aspekty terapii nasledstvennoy gemorragicheskoy teleangiektazii (bolezni Randyu-Oslera) [Modern aspects of hereditary hemorrhagic telangiectasia (Rendu-Osler disease) treatment]. Vrach Skoroy Pomoshchi 2010;12:42-8.
Dupuis-Girod S, Chesnais AL, Ginon I, Dumortier J, Saurin JC, Finet G, et al. Long-term outcome of patients with hereditary hemorrhagic telangiectasia and severe hepatic involvement after orthotopic liver transplantation: A single-center study. Liver Transpl 2010;16(3):340-7. DOI: 10.1002/lt.21990.
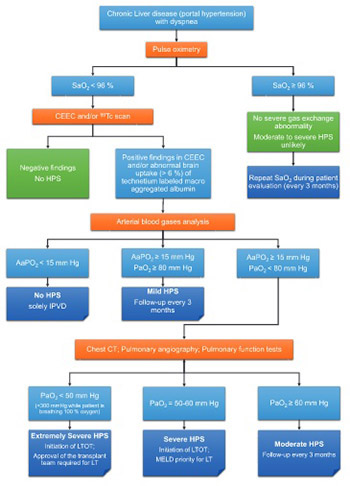
Downloads
Additional Files
Published
How to Cite
Accepted 2017-04-18
Published 2017-11-20





