Retrospective mutational analysis of NPHS1, NPHS2, WT1 and LAMB2 in children with steroid-resistant focal segmental glomerulosclerosis – a single-centre experience
DOI:
https://doi.org/10.17305/bjbms.2014.2270Keywords:
gene variant, NPHS2, WT1, children, steroid-resistant nephrotic syndrome, FSGSAbstract
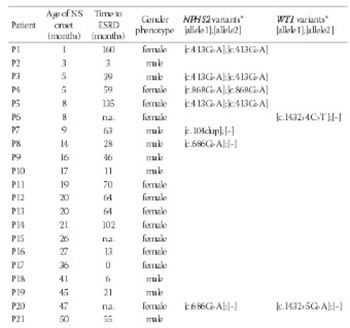
The aim of our study was to examine NPHS1, NPHS2, WT1 and LAMB2 mutations, previously reported in two thirds of patients with nephrotic syndrome with onset before the age of one year old. Genomic DNA samples from Polish children (n=33) with Steroid-ResistantNephrotic Syndrome (SRNS) due to focal segmental glomerulosclerosis (FSGS), manifesting before the age of 13 years old, underwent retrospective analysis of NPHS1, NPHS2, WT1 (exons 8, 9 and adjacent exon/intron boundaries) and LAMB2. No pathogenic NPHS1 or LAMB2 mutations were found in our FSGS cohort. SRNS-causing mutations of NPHS2 and WT1 were detected in 7 of 33 patients (21%), including those with nephrotic syndrome manifesting before one year old: five of seven patients. Four patients had homozygous c.413G>A (p.Arg138Gln) NPHS2 mutations; one subject was homozygous for c.868G>A (p.Val290Met) NPHS2. A phenotypic female had C>T transition at position +4 of the WT1 intron 9 (c.1432+4C>T) splice-donor site, and another phenotypic female was heterozygous for G>A transition at position +5 (c.1432+5G>A). Genotyping revealed a female genotypic gender (46, XX) for the first subject and male (46, XY) for the latter. In addition, one patient was heterozygous for c.104dup (p.Arg36Profs*34) NPHS2; two patients carried a c.686G>A (p.Arg229Gln) NPHS2 non-neutral variant. Results indicate possible clustering of causative NPHS2 mutations in FSGS-proven SRNS with onset before age one year old, and provide additional evidence that patients with childhood steroid-resistant nephrotic syndrome due to focal segmentalglomerulosclerosis should first undergo analysis of NPHS2 coding sequence and WT1 exons 8 and 9 and surrounding exon/intron boundary sequences, followed by gender genotyping.
Citations
Downloads
Downloads
Additional Files
Published
How to Cite
Accepted 2017-07-12
Published 2015-05-20





