De novo mutation in the NOTCH3 gene causing CADASIL
DOI:
https://doi.org/10.17305/bjbms.2014.2297Keywords:
CADASIL, NOTCH3 gene, de novo mutation, CTAbstract
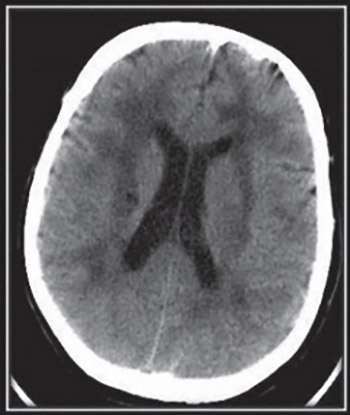
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) is one of the most common hereditary forms of stroke, and migraine with aura, mood disorders and dementia. CADASIL is caused by mutations of the NOTCH3 gene. This mutation is inherited as an autosomal dominant trait. Most individuals with CADASIL have a parent with the disorder. In extremely rare cases, CADASIL may occur due to a spontaneous genetic mutation that occurs for unknown reasons (de novo mutation). We report a new case of patient with de novo mutation of the NOTCH3 gene and a condition strongly suggestive of CADASIL (migraine, stroke, and white matter abnormalities), except that this patient did not have any first-degree relatives with similar symptoms.
Citations
Downloads
Downloads
Additional Files
Published
How to Cite
Accepted 2017-07-20
Published 2014-05-20





