Clinical implications of cellular stress responses
DOI:
https://doi.org/10.17305/bjbms.2012.2510Keywords:
Cell stress, adaptive response, aging, transplantation, cancer, cardiovascular diseaseAbstract
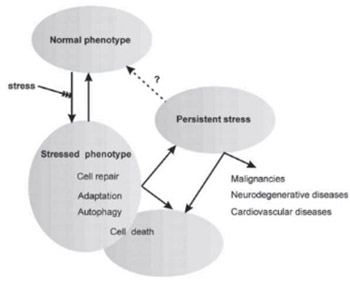
Cellular stress response is a reaction to changes or fluctuations of extracellular conditions that damage the structure and function of macromolecules. Different stressors trigger different cellular responses, namely induce cell repair mechanisms, induce cell responses that result in temporary adaptation to some stressors, induce autophagy or trigger cell death. Inability to repair the damage or exposure to prolonged stress may contribute to aging. Persistent cell stress often enhances susceptibility to cancer and aging associated diseases. Cells and tissues are increasingly being used for transplantations and other novel therapeutic methods in which the quality and well being of cells is of paramount importance for the treatment to succeed. Therefore, discovering the mechanisms of cellular stress responses and the ability to detect and ameliorate them is important in prevention of development of disorders developed by persistent stress and for the success of transplantation and other cell related methods of regenerative medicine.
Citations
Downloads
Downloads
Additional Files
Published
How to Cite
Accepted 2017-09-28
Published 2012-05-20





