Using the BITOLA system to identify candidate genes for Parkinson's disease
DOI:
https://doi.org/10.17305/bjbms.2011.2572Keywords:
BITOLA, Parkinson’s disease, candidate genesAbstract
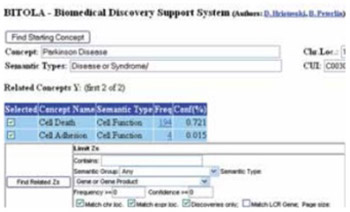
Complexity of multifactorial diseases as Parkinson’s disease (PD) often complicate identifying causal genetic factors by traditional approaches such as positional cloning and candidate gene analyses. PD is etiologically and genetically complex disease and second most common neurodegenerative disorder after Alzheimer’s disease. The most cases of PD are idiopathic and small growing subset of individuals have single gene defect as the cause. The main goal of this research was to identify the potential candidate genes for idiopathic PD by using biomedical discovery support system (BITOLA). For detecting the potential candidate genes for PD was used opened system of bioinformatics tool BITOLA. Data of chromosome location, tissue specific expression of potential candidate genes and their potential association with PD were obtained from Medline, Locus Link, Gene Cards and OMIM. By using BITOLA system is identified 17 genes as potential candidate genes for PD. The role of three genes (MAPT, PARK2, UCHL1) in PD were confirmed earlier. Discovering the novel candidate genes for multifactiorial diseases by using specially mentioned bioinformatics tool BITOLA could offer the new opportunity for researching genetics base of PD without using tissue samples of patients.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
How to Cite
Accepted 2017-10-17
Published 2011-08-20





