Angiosarcoma of the ovary in an 11 year old girl: Case report and review of the literature
DOI:
https://doi.org/10.17305/bjbms.2011.2599Keywords:
primary angiosarcoma, ovary, childhoodAbstract
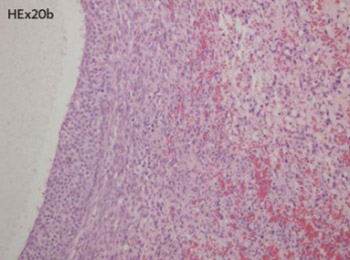
Sarcomas of the female genital tract in general are rare and ovarian sarcomas comprise less than 1% of ovarian malignancies. In the literature there are 15 reported angiosarcomas of patients 21 year old and younger with no one originated in the ovary. We report a case of ovarian angiosarcoma in an 11 year old girl, presented with left side hip pain. MRI of abdomen and pelvis confirmed expansive solid and cystic mass occupied both ovaries. Imunohistochemistry staining was performed, CD34, Factor VIII, CD31, in order to confirm the diagnosis. Final diagnosis was angiosarcoma. The patient received 6 cycles of chemotherapy, according to the CWS-2002P protocol. 8 months after the diagnosis was established, there were no signs of any tumors according to the ultrasound, CT scan, and MRI. Although, extremely rare, angiosarcoma can also affect children and this diagnosis should be considered carefully in tumor with rich vascular network, necrosis and brisk mitotic activity
Citations
Downloads
Additional Files
Published
How to Cite
Accepted 2017-10-23
Published 2011-05-20





