Genetic secrets of long-term glioblastoma survivors
DOI:
https://doi.org/10.17305/bjbms.2018.3717Keywords:
Glioblastoma, genetics, IDH, MGMT, chromosome 1p/19q, long term survival, extreme survivorsAbstract
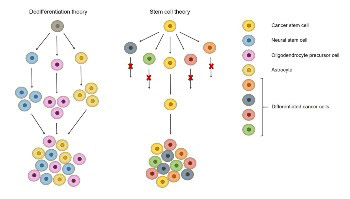
Glioblastomas are the most aggressive and lethal primary astrocytic tumors of the central nervous system. They account for 60% to 70% of all gliomas and the majority are diagnosed in Caucasian male patients at advanced age. Genetic analyses of glioblastoma show a great intra- and inter-tumor heterogeneity, which opens up a debate about its cellular origin. Different types of brain cells, including astrocytes, neural stem cells, oligodendrocyte precursor cells and glioblastoma stem cells are proposed to have a role in tumor initiation and spreading; however, data is still inconclusive. Due to short life expectancy, long-term glioblastoma survivors are defined as patients who live longer than two years post-diagnosis. Extreme survivors, living 10 years or more after diagnosis, comprise less than 1% of all patients. Molecular testing indicates genetic differences between short- and long-term survivors with glioblastoma. The most informative are IDH1/2 gene mutations and MGMT promoter methylation, which are associated with a better response to standard clinical care. Moreover, a decreased expression of the CHI3L1, FBLN4, EMP3, IGFBP2, IGFBP3, LGALS3, MAOB, PDPN, SERPING1 and TIMP1 genes has been associated with prolonged survival. In addition, emerging evidence suggests the role of different microRNAs in predicting patient survival. Other factors that may affect the survival of glioblastoma patients include clinical/demographic characteristics such as seizures at presentation, age at diagnosis, and the extent of surgical resection. Because of the small number of long-term survivors with glioblastoma, comparative studies on genetic differences between short- and long-term survivors are challenging. To improve patient management and clinical outcomes, a thorough “omics” approach is necessary for identifying differences between short- and long-term survivors with glioblastoma.
Citations
Downloads
Downloads
Additional Files
Published
How to Cite
Accepted 2018-07-25
Published 2019-05-20





