EP1 receptor is involved in prostaglandin E2-induced osteosarcoma growth
DOI:
https://doi.org/10.17305/bjbms.2019.4177Keywords:
osteosarcoma, PGE2, EP1, SC51089, prostaglandinAbstract
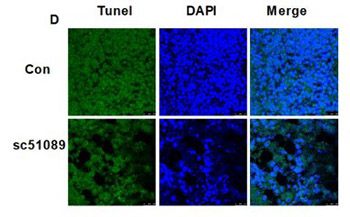
Recent studies showed that the activation of prostaglandin (PG) receptor EP1 promotes cell migration and invasion in different cancers. The aim of this study was to investigate the role of EP1 in the proliferation of osteosarcoma (OS) cells in vitro and in vivo. EP1 mRNA and protein levels were analyzed by real-time RT-PCR and Western blot, respectively in human OS cell lines MG63, OS732, U-2OS, and 143B compared to human fetal osteoblastic hFOB 1.19 cells. MG63 cells were treated with PGE2, EP1 specific agonist 17-PT-PGE2, 17-PT-PGE2 + EP1 specific antagonist SC51089, or DMSO (control). EP1R-siRNA or a non-silencing irrelevant RNA duplex (negative control) were used for the transfection of MG63 cells, followed by PGE2 treatment. Nude mice carrying MG63 xenografts were treated with SC51089 (2 mg/kg/day). MG63 cells/xenografts were analyzed by MTT assay, TUNEL assay, PKC enzyme activity assay, and Western blot (EP1 and apoptotic proteins), and tumor growth/volume was evaluated in mice. EP1 levels were significantly higher in OS cells compared to osteoblasts. PGE2 or 17-PT-PGE2 treatment increased the proliferation and decreased the apoptosis of MG63 cells. Inhibition of EP1 by SC51089 or siRNA markedly decreased the viability of MG63 cells. Similarly, SC51089 treatment significantly inhibited MG63 cell proliferation and promoted apoptosis in vivo. The silencing of EP1 receptor by siRNA or blockade of EP1 signaling by SC51089 activated extrinsic and intrinsic apoptotic pathways both in vivo and in vitro, as evidenced by increased levels of Bax, cyt c, cleaved caspase-3, caspase-8 and caspase-9. EP1 appears to be involved in PGE2-induced proliferative activity of MG63 cells. Antagonizing EP1 may provide a novel therapeutic approach to the treatment of OS.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
How to Cite
Accepted 2019-03-30
Published 2019-08-20





