The natural adaptive evolution of cancer: The metastatic ability of cancer cells
DOI:
https://doi.org/10.17305/bjbms.2019.4565Keywords:
Cancer, metastasis, macrophage–cancer fusion cells, cancer fusion cells, CFCs, hypothesis, reviewAbstract
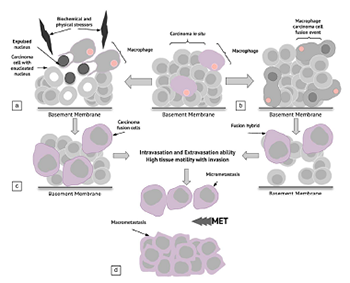
The ability of cancer to adapt renders it one of the most challenging pathologies of all time. It is the most dreaded pathological entity because of its capacity to metastasize to distant sites in the body, and 90% of all cancer-related deaths recorded to date are attributed to metastasis. Currently, three main theories have been proposed to explain the metastatic pathway of cancer: the epithelial–mesenchymal transition (EMT) and mesenchymal–epithelial transition (MET) hypothesis (1), the cancer stem cell hypothesis (2), and the macrophage–cancer cell fusion hybrid hypothesis (3). We propose a new hypothesis, i.e., under the effect of particular biochemical and/or physical stressors, cancer cells can undergo nuclear expulsion with subsequent macrophage engulfment and fusion, with the formation of cancer fusion cells (CFCs). The existence of CFCs, if confirmed, would represent a novel metastatic pathway and a shift in the extant dogma of cancer; consequently, new treatment targets would be available for this adaptive pathology.
Citations
Downloads
Downloads
Additional Files
Published
How to Cite
Accepted 2020-01-19
Published 2020-08-03





