Asymptomatic neurotoxicity of amyloid β-peptides (Aβ1-42 and Aβ25-35) on mouse embryonic stem cell-derived neural cells
DOI:
https://doi.org/10.17305/bjbms.2020.4639Keywords:
Amyloid β-peptides, Alzheimer’s disease, reactive oxygen species, oxidative stress, 46C mouse embryonic stem cellAbstract
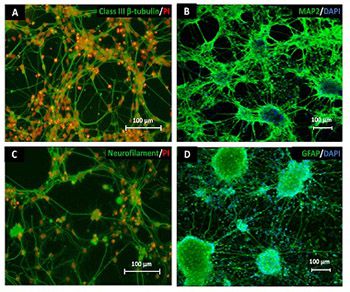
One of the strategies in the establishment of in vitro oxidative stress models for neurodegenerative diseases, such as Alzheimer’s disease (AD), is to induce neurotoxicity by amyloid beta (Aβ) peptides in suitable neural cells. Presently, data on the neurotoxicity of Aβ in neural cells differentiated from stem cells are limited. In this study, we attempted to induce oxidative stress in transgenic 46C mouse embryonic stem cell-derived neurons via treatment with Aβ peptides (Aβ1-42 and Aβ25-35). 46C neural cells were generated by promoting the formation of multicellular aggregates, embryoid bodies in the absence of leukemia inhibitory factor, followed by the addition of all-trans retinoic acid as the neural inducer. Mature neuronal cells were exposed to different concentrations of Aβ1-42 and Aβ25-35 for 24 h. Morphological changes, cell viability, and intracellular reactive oxygen species (ROS) production were assessed. We found that 100 µM Aβ1-42 and 50 µM Aβ25-35 only promoted 40% and 10%, respectively, of cell injury and death in the 46C-derived neuronal cells. Interestingly, treatment with each of the Aβ peptides resulted in a significant increase of intracellular ROS activity, as compared to untreated neurons. These findings indicate the potential of using neurons derived from stem cells and Aβ peptides in generating oxidative stress for the establishment of an in vitro AD model that could be useful for drug screening and natural product studies.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
How to Cite
Accepted 2020-03-04
Published 2021-02-01





