An insight into osteoarthritis susceptibility: Integration of immunological and genetic background
DOI:
https://doi.org/10.17305/bjbms.2020.4735Keywords:
osteoarthritis, immunology, genetic polymorphisms, genetic susceptibilityAbstract
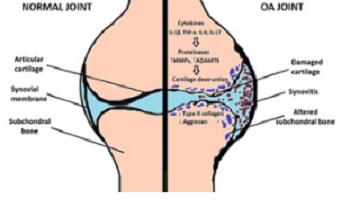
Osteoarthritis (OA) is a progressive degenerative disease that affects all synovial joints, causing the disability of the main locomotor diarthrodial joints. OA pathogenesis is caused by a complex interplay between a number of genetic and environmental risk factors, involved in the early onset and progression of this chronic inflammatory joint disease. Uncovering the underlying immunological and genetic mechanisms will enable an insight into OA pathophysiology and lead to novel and integrative approaches in the treatment of OA patients, together with a reduction of the disease risk, or a delay of its onset in susceptible patients.
Citations
Downloads
Download data is not yet available.
Downloads
Additional Files
Published
01-04-2021
How to Cite
1.
An insight into osteoarthritis susceptibility: Integration of immunological and genetic background. Biomol Biomed [Internet]. 2021 Apr. 1 [cited 2026 Jan. 28];21(2):155-62. Available from: https://www.bjbms.org/ojs/index.php/bjbms/article/view/4735
Received 2020-04-07
Accepted 2020-09-11
Published 2021-04-01
Accepted 2020-09-11
Published 2021-04-01





