Stomach-specific c-Myc overexpression drives gastric adenoma in mice through AKT/mammalian target of rapamycin signaling
DOI:
https://doi.org/10.17305/bjbms.2020.4978Keywords:
c-Myc, gastric adenoma, transgenic, AKT/mammalian target of rapamycinAbstract
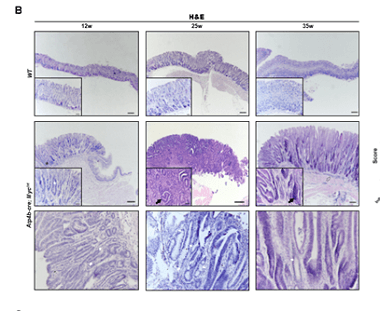
Gastric cancer (GC) is one of the most common malignant cancers in the world. c-Myc, a well-known oncogene, is commonly amplified in many cancers, including gastric cancer. However, it is still not completely understood how c-Myc functions in GC. Here, we generated a stomach-specific c-Myc transgenic mouse model to investigate its role in GC. We found that overexpression of c-Myc in Atp4b+ gastric parietal cells could induce gastric adenoma in mice. Mechanistically, c-Myc promoted tumorigenesis via the AKT/mTOR pathway. Furthermore, AKT inhibitor (MK-2206) or mTOR inhibitor (Rapamycin) inhibited the proliferation of c-Myc overexpressing gastric cancer cell lines. Thus, our findings highlight that gastric tumorigenesis can be induced by c-Myc overexpression through activation of the AKT/mTOR pathway.
Citations
Downloads
Downloads
Additional Files
Published
How to Cite
Accepted 2020-11-16
Published 2021-08-01





