The role of glycogen synthase kinase 3 (GSK3) in cancer with emphasis on ovarian cancer development and progression: A comprehensive review
DOI:
https://doi.org/10.17305/bjbms.2020.5036Keywords:
GSK3, ovarian cancer, therapeutic targetAbstract
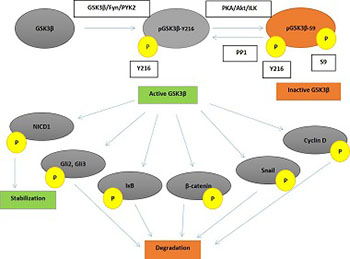
Glycogen synthase kinase 3 (GSK3) is a monomeric serine-threonine kinase discovered in 1980 in a rat skeletal muscle. It has been involved in various cellular processes including embryogenesis, immune response, inflammation, apoptosis, autophagy, wound healing, neurodegeneration, and carcinogenesis. GSK3 exists in two different isoforms, GSK3α and GSK3β, both containing seven antiparallel beta-plates, a short linking part and an alpha helix, but coded by different genes and variously expressed in human tissues. In the current review, we comprehensively appraise the current literature on the role of GSK3 in various cancers with emphasis on ovarian carcinoma. Our findings indicate that the role of GSK3 in ovarian cancer development cannot be decisively determined as the currently available data support both prooncogenic and tumor-suppressive effects. Likewise, the clinical impact of GSK3 expression on ovarian cancer patients and its potential therapeutic implications are also limited. Further studies are needed to fully elucidate the pathophysiological and clinical implications of GSK3 activity in ovarian cancer.
Citations
Downloads