A novel homozygous exon2 deletion of TRIM32 gene in a Chinese patient with sarcotubular myopathy: A case report and literature review
DOI:
https://doi.org/10.17305/bjbms.2020.5288Keywords:
Sarcotubular myopathy, TRIM32, vacuoles, homozygous deletion, case reportAbstract
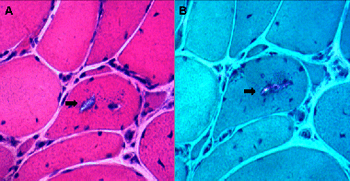
Sarcotubular myopathy (STM) is a rare autosomal recessive myopathy caused by TRIM32 gene mutations. It is predominantly characterized by the weakness of the proximal limb and mild to moderate elevation of creatine kinase (CK) levels. In this study, we describe a 50-year-old Chinese man who exhibited a proximal-to-distal weakness in the muscles of the lower limbs and who had difficulty standing up from a squat position. The symptoms gradually became more severe. He denied a history of cognitive or cardiological problems. The patient’s parents and children were healthy. Histopathological examination revealed dystrophic changes and irregular slit-shaped vacuoles containing amorphous materials. Whole-exome sequencing consisting of protein-encoding regions of 19,396 genes was performed, the results of which identified one novel homozygous 2kb deletion chr9.hg19: g.119460021_119461983del (exon2) in the TRIM32 gene. This was confirmed at the homozygous state with quantitative real-time PCR. Here, we present a Chinese case of STM with one novel mutation in TRIM32 and provide a brief summary of all known pathogenic mutations in TRIM32.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
How to Cite
Accepted 2020-12-18
Published 2021-08-01





