Altered molecular pathways and prognostic markers in active systemic juvenile idiopathic arthritis: integrated bioinformatic analysis
DOI:
https://doi.org/10.17305/bjbms.2021.6016Keywords:
Systemic juvenile idiopathic arthritis, hub genes, neutrophil, prognostic markerAbstract
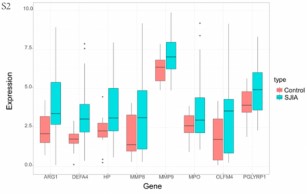
Systemic juvenile idiopathic arthritis (SJIA) is a severe childhood-onset inflammatory disease characterized by arthritis accompanied by systemic auto-inflammation and extra-articular symptoms. While recent advances have unraveled a range of risk factors, the pathomechanisms involved in SJIA and potential prognostic markers for treatment success remain partly unknown. In this study, we included 70 active SJIA and 55 healthy control patients from the National Center for Biotechnology Information to analyze for differentially expressed genes (DEGs) using R. Functional enrichment analysis, protein-protein interaction (PPI), and gene module construction were performed for DEGs and hub gene set. We additionally examined immune system cell composition with CIBERSORT and predicted prognostic markers and potential treatment drugs for SJIA. In total, 94 upregulated and 24 downregulated DEGs were identified. Two specific modules of interest and eight hub genes (ARG1, DEFA4, HP, MMP8, MMP9, MPO, OLFM4, PGLYRP1) were screened out. Functional enrichment analysis suggested that complex neutrophil-related functions play a decisive role in the disease pathogenesis. CIBERSORT indicated neutrophils, M0 macrophages, CD8+ T cells, and naïve B cells to be relevant drivers of disease progression. Additionally, we identified TPM2 and GZMB as potential prognostic markers for treatment response to canakinumab. Moreover, sulindac sulfide, (-)-catechin, and phenanthridinone were identified as promising treatment agents. This study provides a new insight into molecular and cellular pathogenesis of active SJIA and highlights potential targets for further research.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
License
Copyright (c) 2021 Yi Ren, Hannah Labinsky, Andriko Palmowski, Henrik Bäcker, Michael Müller, Arne Kienzle

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2021-08-07
Published 2022-04-01





