Small cell variant of chromophobe renal cell carcinoma: Clinicopathologic and molecular-genetic analysis of 10 cases.
DOI:
https://doi.org/10.17305/bjbms.2021.6935Keywords:
kidney, chromophobe renal cell carcinoma, small cell variantAbstract
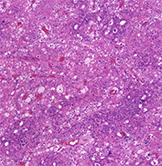
The morphologic diversity of chromophobe renal cell carcinoma (ChRCC) is well-known. Aside from typical morphology, pigmented adenomatoid, multicystic and papillary patterns have been described. Ten cases of CHRCC composed of small cell population in various percentages were analysed, using morphologic parameters, immunohistochemistry and next-generation sequencing (NGS) testing. Patients were five males and five females, with age ranging from 40 to 78years. The size of tumors ranged from 2.2 cm to 11 cm (mean 5.17 cm). Small cell component comprised 10 to 80% of the tumor volume, while the remaining was formed by cells with classic ChRCC morphology. The immunohistochemical profile of the small cell component was consistent with typical ChRCC immunophenotype, with CD117 and CK7 positivity. Neuroendocrine markers were negative. Mutations of 13 genes were found: DCIER1, FGFR3, JAK3, SUFO, FAM46C, FANCG, MET, PLCG2, APC, POLE, EPICAM, MUTYH and AR. However, only the PLCG2 mutation is considered pathogenic.The small cell variant of ChRCC further highlights and expand upon existing morphologic heterogeneity spectrum. Recognition of small cell variant of CHRCC is not problematic in tumors, where the “classic” CHRCC component is present. However, in limited material (i.e., core biopsy), this may present a diagnostic challenge. Based on the limited follow-up data available, it appears that the small cell tumor component had no impact on prognosis, since there was no aggressive behavior documented. Awareness of this unusual pattern and applying additional sections to find classic morphology of ChRCC, as well as excluding neuroendocrine nature by immunohistochemistry, may help resolve difficult cases.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
License
Copyright (c) 2022 Joanna Rogala, Fumiyoshi Kojima, Reza Alaghehbandan, Nikola Ptakova, Ana Bravc, Stela Bulimbasic, Delia Perez Montiel, Maryna Slisarenko, Leila Ali, Levente Kuthi, Kristyna Pivovarcikova, Kvetoslava Michalova, Boris Bartovic, Adriena Bartos Vesela, Olga Dolejsova, Michal Michal, Ondrej Hes

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2022-01-31
Published 2022-07-29





