Integrated transcriptomic and proteomic analysis reveals potential targets for heart regeneration
DOI:
https://doi.org/10.17305/bjbms.2022.7770Keywords:
Proteomic, transcriptomic, heart regeneration, myocardial infarctionAbstract
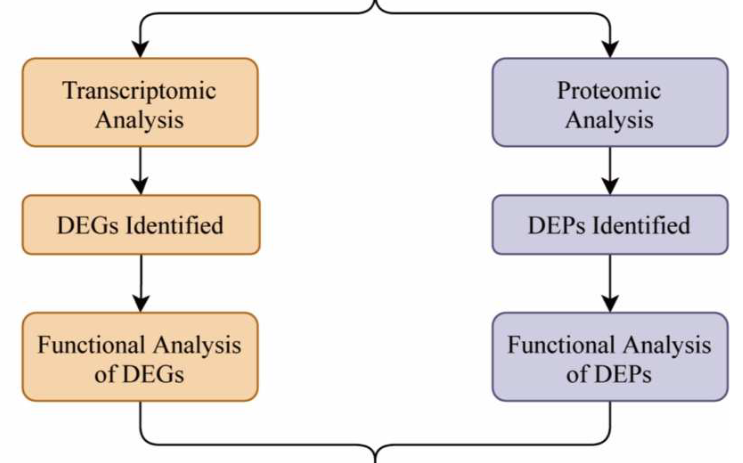
Research on the regenerative capacity of the neonatal heart could open new avenues for the treatment of myocardial infarction (MI). However, the mechanism of cardiac regeneration remains unclear. In the present study, we constructed a mouse model of heart regeneration and then performed transcriptomic and proteomic analyses on them. Gene Ontology (GO) enrichment analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, and Gene Set Enrichment Analysis (GSEA) of differentially expressed genes (DEGs) were conducted. Western blot (WB) and qPCR analyses were used to validate the hub genes expression. As a result, gene expression at the mRNA level and protein level is not the same. We identified 3186 DEGs and 42 differentially expressed proteins (DEPs). Through functional analysis of DEGs and DEPs, we speculate that biological processes such as ubiquitination, cell cycle, and oxygen metabolism are involved in heart regeneration. Integrated transcriptomic and proteomic analysis identified 19 hub genes and Ankrd1, Gpx3, and Trim72 were screened out as potential regulators of cardiac regeneration through further expression verification. In conclusion, we combined transcriptomic and proteomic analyses to characterize the molecular features during heart regeneration in neonatal mice. Finally, Ankrd1, Gpx3, and Trim72 were identified as potential targets for heart regeneration therapy.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
License
Copyright (c) 2022 Liu Liu, Tongtong Yang, Qiqi Jiang, Jiateng Sun, Lingfeng Gu, Sibo Wang, Yafei Li, Bingrui Chen, Di Zhao, Rui Sun, Qiming Wang, Hao Wang, Liansheng Wang

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2022-08-03
Published 2023-01-06





