Deletion of sphingosine kinase 2 attenuates cigarette smoke-mediated chronic obstructive pulmonary disease-like symptoms by reducing lung inflammation
DOI:
https://doi.org/10.17305/bjbms.2022.8034Keywords:
SphK2, COPD, CFTR, S1P, pulmonary inlammationAbstract
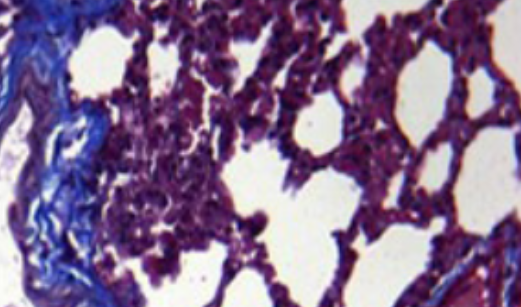
Cigarette smoke (CS) is the leading cause of chronic obstructive pulmonary disease (COPD), which is characterized by chronic bronchial inflammation and emphysema. Growing evidence supports the hypothesis that dysfunctional cystic fibrosis transmembrane conductance regulator (CFTR) is critically involved in the pathogenesis of CS-mediated COPD. However, the underlying mechanism remains unclear. Here, we report that supressed CFTR expression is strongly associated with abnormal phospholipid metabolism and increased pulmonary inflammation. In a CS-exposed mouse model with COPD-like symptoms, we found that pulmonary expression of sphingosine kinase 2 (SphK2) and sphingosine-1-phosphate (S1P) secretion were significantly upregulated. Therefore, we constructed a SphK2 gene knockout (SphK2-/-) mouse. After CS exposure for six months, histological lung section staining showed disorganized alveolar structure, increased pulmonary fibrosis, and emphysema-like symptoms in wild-type (WT) mice, which were less pronounced in SphK2-/- mice. Further, SphK2 deficiency also decreased CS-induced pulmonary inflammation, which was reflected by a remarkable reduction in pulmonary infiltration of CD45+CD11b+ neutrophils subpopulation and low levels of IL-6 and IL-33 in bronchial alveolar lavage fluid. However, treatment with S1P receptor agonist suppressed CFTR expression and increased Nf-κB-p65 expression and its nuclear translocation in CS-exposed SphK2-/-mice, which also aggravated small airways fibrosis and pulmonary inflammation. In contrast, inhibition of S1P signaling with the S1P receptor analogue FTY720 rescued CFTR expression, suppressed Nf-κB-p65 expression and nuclear translocation, and alleviated pulmonary fibrosis and inflammation after CS exposure. Our results demonstrate that SphK2-mediated S1P production plays a crucial role in the pathogenesis of CS-induced COPD-like disease by impairing CFTR activity and promoting pulmonary inflammation and fibrosis.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
License
Copyright (c) 2022 Yanhui Chen, Yongrong Zhang, Cheng Rao, Jieyun Huang, Qiong Qing

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2022-10-03
Published 2023-03-16





