The role of Rapsyn in neuromuscular junction and congenital myasthenic syndrome
DOI:
https://doi.org/10.17305/bb.2022.8641Keywords:
Rapsyn, neuromuscular junction (NMJ), congenital myasthenic syndrome (CMS), mutation, medication, central nervous system (CNS)Abstract
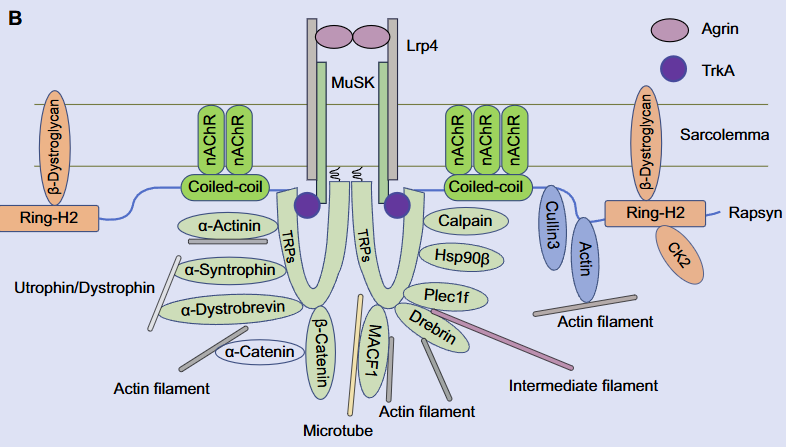
Rapsyn, an intracellular scaffolding protein associated with the postsynaptic membranes in the neuromuscular junction (NMJ), is critical for nicotinic acetylcholine receptor clustering and maintenance. Therefore, Rapsyn is essential to the NMJ formation and maintenance, and Rapsyn mutant is one of the reasons causing the pathogenies of congenital myasthenic syndrome (CMS). In addition, there is little research on Rapsyn in the central nervous system (CNS). In this review, the role of Rapsyn in the NMJ formation and the mutation of Rapsyn leading to CMS will be reviewed separately and sequentially. Finally, the potential function of Rapsyn is prospected.
Citations
Downloads
Downloads
Additional Files
Published
License
Copyright (c) 2023 Xufeng Liao, Yingxing Wang, Xinsheng Lai, Shunqi Wang

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2023-02-17
Published 2023-09-04





