The role of chidamide in the treatment of B-cell non-Hodgkin lymphoma: An updated systematic review
DOI:
https://doi.org/10.17305/bb.2023.8791Keywords:
Chidamide, tucidinostat, histone deacetylase (HDAC), histone deacetylase inhibitors, non-Hodgkin lymphoma (NHL)Abstract
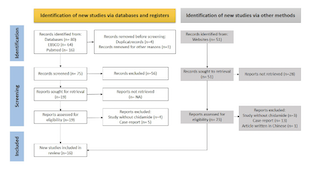
B-cell non-Hodgkin lymphoma (B-NHL) is a lymphoid malignancy derived from B-cells that remains difficult to treat. Moreover, relapses and refractory cases are common. Abnormalities in epigenetic mechanisms, such as imbalanced histone acetylation affecting certain genes, contribute to relapses and refractory cases. Chidamide (tucidinostat) is a novel histone deacetylase inhibitor that can reverse this epigenetic imbalance and has been approved for the treatment of T-cell malignancies. However, the use of chidamide for B-NHL remains limited, and the lack of relevant literature exacerbates this limitation. We conducted this review to summarize the anticancer activity of chidamide against B-NHL and its clinical applications to overcome drug resistance. This systematic review was conducted according to the PRISMA 2020 guidelines, using some keyword combinations from MEDLINE and EBSCO. The inclusion and exclusion criteria were also defined. Of the 131 records retrieved from databases, 16 were included in the review. Nine articles revealed that chidamide limited tumor progression by modifying the tumor microenvironment, stopping the cell cycle, inducing apoptosis and autophagy, and enhancing complement-dependent and antibody-dependent cell-mediated cytotoxicities.According to seven other studies, administering chidamide in combination with another existing therapeutic regimen may benefit not only patients with relapsed/refractory B-NHL, but also those with newly diagnosed B-NHL. Chidamide plays many important roles in limiting B-NHL progression through epigenetic modifications. Thus, combining chidamide with other anticancer drugs may be more beneficial for patients with newly diagnosed and relapsed/refractory B-NHL.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
License
Copyright (c) 2023 Hastono Ridwansyah, Indra Wijaya, Muhammad Hasan Bashari, Achmad Hussein Sundawa Kartamihardja, Bethy Suryawathy Hernowo

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2023-03-26
Published 2023-09-04





