Lipoprotein(a): Role in atherosclerosis and new treatment options
DOI:
https://doi.org/10.17305/bb.2023.8992Keywords:
Lipoprotein(a) (Lp(a)), lipids, atherosclerosis, major adverse cardiovascular events (MACE), cardiovascular (CV) diseases, cardiovascular preventionAbstract
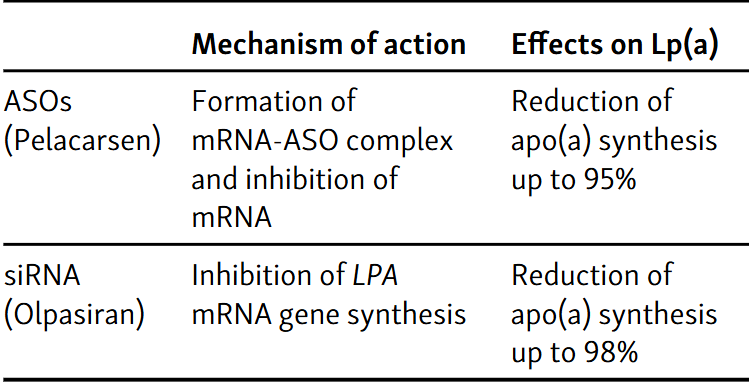
Atherosclerosis is a chronic process characterized by inflammation and the progressive accumulation of inflammatory cells and lipids in the blood vessel wall, resulting in narrowing of the blood vessel’s circumference. Treatment of people with dyslipidemia aims to reduce the risk of developing atherosclerotic disease and prevent major adverse cardiovascular events (MACE). The results of previous studies indicated that lipoprotein(a) (Lp(a)) is a critical causal factor in the estimated risk of developing a cardiovascular (CV) incident even after achieving desirable low-density lipoprotein (LDL) cholesterol levels. Lp(a) is a low-density lipoprotein particle, like LDL cholesterol. The levels of Lp(a) in plasma are genetically determined. Lp(a) catabolism is still controversial. The pathogenic potential of Lp(a) can be divided into three categories: promotion of plaque formation, thrombogenicity, and proinflammatory effects. Lp(a) levels above the 75th percentile reduced the risk of aortic valve stenosis and myocardial infarction, whereas higher levels (above 90th percentile) were associated with an increased risk of heart failure. However, no hypolipidemic agents have been approved for targeted use in patients with high Lp(a) levels. There are insufficient randomized controlled trials assessing CV outcomes that would support the evidence that current treatment options, which effectively lower Lp(a) levels, also effectively prevent CV event. However, according to some studies, there is strong evidence that better CV outcome is one of the benefits of such therapy. The results of ongoing clinical trials are eagerly awaited.
Citations
Downloads
Downloads
Additional Files
Published
License
Copyright (c) 2023 Dragana Tomic Naglic, Mia Manojlovic, Sladjana Pejakovic, Kristina Stepanovic, Jovana Prodanovic Simeunovic

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2023-04-13
Published 2023-07-03





