Diagnostics of common microdeletion syndromes using fluorescence in situ hybridization: single center experience in a developing country
DOI:
https://doi.org/10.17305/bjbms.2016.994Keywords:
Microdeletion syndrome, DiGeorge, Williams, Prader-Willi, Angelman, Wolf-Hirschhorn, Fluorescent in situ hybridizationAbstract
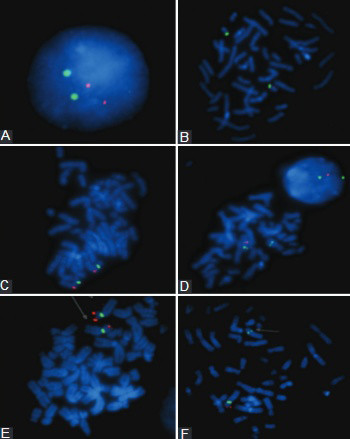
Microdeletion syndromes are caused by chromosomal deletions of less than 5 megabases which can be detected by fluorescence in situ hybridization (FISH). We evaluated the most commonly detected microdeletions for the period from June 01, 2008 to June 01, 2015 in the Federation of Bosnia and Herzegovina, including DiGeorge, Prader-Willi/Angelman, Wolf-Hirschhorn, and Williams syndromes. We report 4 patients with DiGeorge syndromes, 4 patients with Prader-Willi/Angelman, 4 patients with Wolf-Hirschhorn syndrome, and 3 patients with Williams syndrome in the analyzed 7 year period. Based on the positive FISH results for each syndrome, the incidence was calculated for the Federation of Bosnia and Herzegovina. These are the first reported frequencies of the microdeletion syndromes in the Federation of Bosnia and Herzegovina.
Citations
Downloads
References
Dallapiccola B, Mingarelli R, Novelli G. The link between cytogenetics and mendelism. Biomed &Pharmacother 1995; 49:83-93.
http://dx.doi.org/10.1016/0753-3322(96)82592-3
Hunter AG. Outcome of the routine assessment of patients with mental retardation in a genetics clinic. Am J Med Genet 2000; 90:60–8.
http://dx.doi.org/10.1002/(SICI)1096-8628(20000103)90:1<60::AID-AJMG11>3.0.CO;2-P
Kirchhoff M, Bisgaard AM, Bryndorf T, Gerdes T. MLPA analysis for a panel of syndromes with mental retardation reveals imbalances in 5.8% of patients with mental retardation and dysmorphic features, including duplications of the Sotos syndrome and Williams-Beuren syndrome regions. Eur J Med Genet 2007; 50:33–42.
http://dx.doi.org/10.1016/j.ejmg.2006.10.002
Flint J, Wilkie AO, Buckle VJ, Winter RM, Holland AJ, McDermid HE. The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat Genet 1995; 9:132–40. http://dx.doi.org/10.1038/ng0295-132
Vissers LE, De Vries BB, Osoegawa K, Janssen IM, Feuth T, Choy CO, et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet 2003; 73:1261–70.
http://dx.doi.org/10.1086/379977
Tézenas Du, Montcel S, Mendizabai H, Aymé S, Lévy A, Philip N. Prevalence of 22q11 microdeletion. J Med Genet 1996; 33:719. http://dx.doi.org/10.1136/jmg.33.8.719
Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child 1998; 79:348-51. http://dx.doi.org/10.1136/adc.79.4.348
Oskarsdóttir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in western Sweden. Arch Dis Child 2004; 89:148–151.
http://dx.doi.org/10.1136/adc.2003.026880
Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet 1998; 35:789–790.
http://dx.doi.org/10.1136/jmg.35.9.789-a
Schinzel A. Catalogue of unbalanced chromosome aberrations in man. 2nd ed. New York: W de Gruyter; 2001.
Jones KL. Smith's recognizable patterns of human malformation. 6th ed. Philadelphia: Elsevier Saunders; 2006.
Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletion: a European collaborative study. J Med Genet 1997; 34:798-4.
http://dx.doi.org/10.1136/jmg.34.10.798
Chen KS, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault AC,et al. Homologous recombination of a flaking repeat gene cluster is a mechanism for a common continuous gene deletion syndrome. Nat Genet 1997; 17:154-63.
http://dx.doi.org/10.1038/ng1097-154
Shaw CJ, Bi W, Lupski RJ. Genetic proof of unequal meiotic crossover in reciprocal deletion and duplication of 17p11.2. Am J Hum Genet 2002; 71:1072-81.
http://dx.doi.org/10.1086/344346
Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet 2009; 1:3-13.
http://dx.doi.org/10.1038/ejhg.2008.165
Vogels A, Fryns PJ. The Prader-Willi syndrome and the Angelman Syndrome. Genetic Counsel 2002; 13:385-96.
Battaglia A, Carey JC, Wright TJ. Wolf-Hirschhorn (4p-) syndrome. Adv Pediatr 2001; 48: 75–113.
Battaglia A, Filippi T, Carey JC. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet 2008; 148(4):246-51.
http://dx.doi.org/10.1002/ajmg.c.30187
Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, et al. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet 2008; 148C: 257-269.
http://dx.doi.org/10.1002/ajmg.c.30190
Battaglia A, Carey JC, South ST, Wright TJ. Wolf-Hirschhorn syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K (eds). Gene Reviews [Internet]. Seattle: University of Washington; 2010[cited 14 July 2015]. Available from NCBI Bookshelf: http://www.ncbi.nlm.nih.gov/books/NBK1183/
Downloads
Additional Files
Published
How to Cite
Accepted 2016-01-25
Published 2016-03-03





