Epigenetic alterations of the Wnt signaling pathway in cancer: a mini review
DOI:
https://doi.org/10.17305/bjbms.2014.4.205Keywords:
Cancer, epigenetics, Wnt signaling pathwayAbstract
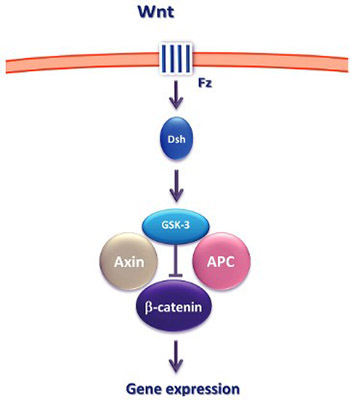
Epigenetic mechanisms play a crucial role in cellular proliferation, migration and differentiation in both normal and neoplastic development. One of the key signaling pathways whose components are altered through the epigenetic mechanisms is the Wnt signaling pathway. In this review, we briefly discuss the key concepts of epigenetics and focus on the recent advances in the Wnt signaling pathway research and its potential diagnostic and therapeutic implications.
Citations
Downloads
Download data is not yet available.
Downloads
Additional Files
Published
12-11-2014
How to Cite
1.
Epigenetic alterations of the Wnt signaling pathway in cancer: a mini review. Biomol Biomed [Internet]. 2014 Nov. 12 [cited 2026 Jan. 28];14(4):191-4. Available from: https://www.bjbms.org/ojs/index.php/bjbms/article/view/a191
Received 2014-11-01
Accepted 2014-11-02
Published 2014-11-12
Accepted 2014-11-02
Published 2014-11-12





