Silence of p15 expression by RNAi enhances cisplatin resistance in hepatocellular carcinoma cells
DOI:
https://doi.org/10.17305/bjbms.2012.2523Keywords:
hepatocellular carcinoma, cell cycle, p15, RNA interference, drug resistanceAbstract
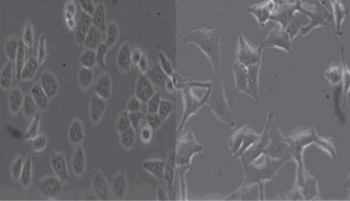
The insensitivity of hepatocellular carcinoma to chemotherapy is associated with alternation in tumor cell cycling. This current study was designed to investigate the impact of p15 silencing on the sensitivity of Human hepatocellular carcinoma HepG2 cells to cisplatin. HepG2/CDDP/1.6 and HepG2/CDDP/2.0 cells were induced by culture with increased doses of cisplatin and their sensitivities to cis-Diamine dichloroplatinum (CDDP) were determined by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). The impacts of p15 silencing on the cell cycling and P-gp expression were characterized by flow cytometry, RT-PCR and Western blot assays, respectively Knockdown of p15 expression dramatically reduced the relative levels of p15 expression and the frequency of phase G1, promoting cell cycling. On the other hand, knockdown of p15 expression significantly up-regulated the expression of P-glycoprotein (P-gp) in HepG2/CDDP/2.0 cells, associated with the increased resistance of HepG2 cells to CDDP in vitro.
In conclusion, the p15 may be a critical regulator of the development of CDDP resistance in HepG2 cells.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
How to Cite
Accepted 2017-10-02
Published 2012-02-20





