Combined inhibition of ACK1 and AKT shows potential toward targeted therapy against KRAS-mutant non-small-cell lung cancer
DOI:
https://doi.org/10.17305/bjbms.2020.4746Keywords:
KRAS mutation, NSCLC, drug combination, Chou-Talalay, synergistic effectAbstract
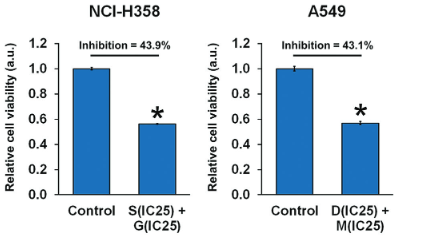
Non-small-cell lung cancer (NSCLC) with Kirsten RAt Sarcoma 2 viral oncogene homolog (KRAS) mutation has become a clinical challenge in cancer treatment as KRAS-mutant tumors are often resistant to conventional anti-tumor therapies. Activated CDC42-associated kinase 1 (ACK1), an activator of protein kinase B (AKT), is a promising target for KRAS-mutant tumor therapy, but the downstream ACK1 signaling remains poorly understood. The aim of this study was to evaluate the effectiveness of combined ACK1/AKT inhibition on the proliferation, migration, invasion, and apoptosis of KRAS-mutant NSCLC cell lines (NCI-H23, NCI-H358, and A549). The cells were treated with an inhibitor of either ACK1 (dasatinib or sunitinib) or AKT (MK-2206 or GDC-0068), and the optimal concentrations of the two yielding synergistic tumor-killing effects were determined by applying the Chou-Talalay equation for drug combinations. We showed that combined administration of ACK1 and AKT inhibitors at the optimal concentrations effectively suppressed NSCLC cell viability and promoted apoptosis while inducing cell cycle arrest at the G2 phase. Moreover, NSCLC cell migration and invasion were inhibited by combined ACK1/AKT inhibition. These phenomena were associated with the reduced phosphorylation levels of ACK1 and AKT (at Ser473 and Thr308), as well as alterations in caspase-dependent apoptotic signaling. Collectively, our results demonstrate the promising therapeutic potential of combined ACK1/AKT inhibition as a strategy against KRAS-mutant NSCLC. Our findings provide the basis for the clinical translation of biological targeted drugs (ACK1 and AKT inhibitors) and their rational combination in cancer treatment.
Citations
Downloads
Downloads
Additional Files
Published
Issue
Section
Categories
License
Copyright (c) 2020 Xiangjing Yu

This work is licensed under a Creative Commons Attribution 4.0 International License.
How to Cite
Accepted 2020-06-01
Published 2021-04-01





