An insight into lung cancer: a comprehensive review exploring ALK TKI and mechanisms of resistance
DOI:
https://doi.org/10.17305/bjbms.2021.5859Keywords:
NSCLC, anaplastic lymphoma kinase, ALK, tyrosine kinase inhibitors, TKI, resistance mechanismsAbstract
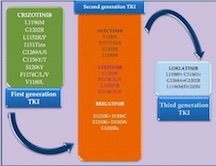
Implementation of precision medicine in lung cancer has benefited from intense research in the past years, developing subsequently an improved quality of life and increased overall survival of the patients. Targeted therapy has become one of the most important therapeutic innovations for the non-small cell lung cancer (NSCLC) category with anaplastic lymphoma kinase (ALK) gene rearrangement. The aim of this review is to provide a through overview of the main molecules of ALK tyrosine kinase inhibitors (TKI) with their general and particular mechanisms of resistance, the main methods of ALK gene detection, each with advantages and limits and the future perspectives currently under research which try to overcome the mechanisms of resistance. We have used two of the most reliable medical databases EMBASE and PubMed to properly select the latest and the most relevant articles for this topic. Encouraged by the promising results, the clinical practice was enriched by the approval of tyrosine kinase inhibitor molecules, three generations being developed, each one with more powerful agents than the previous ones. Unfortunately, the resistance to TKI eventually occurs and it may be induced by several mechanisms, either known or unknown. Crizotinib was the most intensely studied TKI , becoming the first molecule approved into clinical practice and although four other drugs have been broadly used (alectinib, ceritinib, brigatinib and lorlatinib) it seems that even the most recently developed one remains imperfect due to the resistance mutations that developed. There are two types of resistance generally described for the entire class and for the particular drugs, but half of them remain unknown
Citations
Downloads
Downloads
Additional Files
Published
How to Cite
Accepted 2021-05-05
Published 2022-02-01





