Remifentanil-induced preconditioning has cross-talk with A1 and A2B adenosine receptors in ischemic-reperfused rat heart
DOI:
https://doi.org/10.17305/bjbms.2016.738Keywords:
Adenosine, Cross-talk, Remifentanil, Reperfusion, PreconditioningAbstract
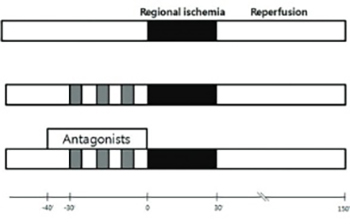
The purpose of this study was to determine whether there is a cross-talk between opioid receptors (OPRs) and adenosine receptors (ADRs) in remifentanil preconditioning (R-Pre) and, if so, to investigate the types of ADRs involved in the cross-talk. Isolated rat hearts received 30 min of regional ischemia followed by 2 hr of reperfusion. OPR and ADR antagonists were perfused from 10 min before R-Pre until the end of R-Pre. The heart rate, left ventricular developed pressure (LVDP),velocity of contraction (+dP/dtmax), and coronary flow (CF) were recorded. The area at risk and area of necrosis were measured. After reperfusion, the LVDP, +dP/dtmax,and CF showed a significant increase in the R-Pre group compared with the control group (no intervention before or after regional ischemia). These increases in the R-Pre group were blocked by naloxone, a nonspecific ADR antagonist, an A1 ADR antagonist, and an A2B ADR antagonist. The infarct size was reduced significantly in the R-Pre group compared with the control group. The infarct-reducing effect in the R-Pre group was blocked by naloxone, the nonspecific ADR antagonist, the A1 ADR antagonist, and the A2B ADR antagonist. The results of this study demonstrate that there is cross-talk between ADRs and OPRs in R-Pre and that A1 ADR and A2B ADR appear to be involved in the cross-talk.
Citations
Downloads
References
Zhang Y, Irwin MG, Wong TM. Remifentanil preconditioning protects against ischemic injury in the intact rat heart. Anesthesiology 2004;101(4):918-923.
http://dx.doi.org/10.1097/00000542-200410000-00017
Kim HS, Kim SY, Kwak YL, Hwang KC, Shim YH. Hyperglycemia attenuates myocardial preconditioning of remifentanil. J Surg Res 2012;174(2):231-237.
http://dx.doi.org/10.1016/j.jss.2011.01.018
Rork TH, Wallace KL, Kennedy DP, Marshall MA, Lankford AR, Linden J. Adenosine A2A receptor activation reduces infarct size in the isolated, perfused mouse heart by inhibiting resident cardiac mast cell degranulation. Am J Physiol Heart Circ Physiol 2008;295:H1825–1833. 10.1152/ajpheart.495.2008.-Mast:
http://dx.doi.org/10.1152/ajpheart.495.2008.-Mast.
Xi J, McIntosh R, Shen X, Lee S, Chanoit G, Criswell H, et al. Adenosine A2A and A2B receptors work in concert to induce a strong protection against reperfusion injury in rat hearts. J Mol Cell Cardiol 2009;47(5):684-690.
http://dx.doi.org/10.1016/j.yjmcc.2009.08.009
Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev 2007;12(3-4):181-188.
http://dx.doi.org/10.1007/s10741-007-9025-2
Lee JW, Joshi S, Chan JS, Wong YH. Differential coupling of μ-, δ-, and κ-opioid receptors to Gα16-mediated stimulation of phospholipase C. J Neurochem 1998;70:2203-2211.
http://dx.doi.org/10.1046/j.1471-4159.1998.70052203.x
Zhang Y, Irwin MG, Wong TM, Chen M, Cao CM. Remifentanil preconditioning confers cardioprotection via cardiac κ- and δ-opioid receptors. Anesthesiology 2005;102:371-378.
http://dx.doi.org/10.1097/00000542-200502000-00020
Peart JN, Gross GJ. Adenosine and opioid receptor-mediated cardioprotection in the rat: evidence for cross-talk between receptors. Am J Physiol Heart Circ Physiol 2003;285(1):H81-89.
http://dx.doi.org/10.1152/ajpheart.00985.2002
Parsons M, Young L, Lee JE, Jacobson KA, Liang BT. Distinct cardioprotective effects of adenosine mediated by differential coupling of receptor subtypes to phospholipases C and D. FASEB J 2000;14:1423-1431.
http://dx.doi.org/10.1096/fj.14.10.1423
Peart J, Willems L, Headrick JP. Receptor and non-receptor-dependent mechanisms of cardioprotection with adenosine. Am J Physiol Heart Circ Physiol 2003;284:H519-527.
http://dx.doi.org/10.1152/ajpheart.00717.2002
Kato R, Ross S, Foëx P. Fentanyl protects the heart against ischaemic injury via opioid receptors, adenosine A1 receptors and KATP channel linked mechanisms in rats. Br J Anaesth 2000;84:204-214.
http://dx.doi.org/10.1093/oxfordjournals.bja.a013404
Chen Z, Li T, Zhang B. Morphine postconditioning protects against reperfusion injury in the isolated rat hearts. J Surg Res 2008;145(2):287-294.
http://dx.doi.org/10.1016/j.jss.2007.07.020
Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE. Inhibiting protease-activated receptor 4 limits myocardial ischemia/reperfusion injury in rat hearts by unmasking adenosine signaling. J Pharmacol Exp Ther 2008;324(3):1045-1054.
http://dx.doi.org/10.1124/jpet.107.133595
Ebrahimi S, Faghihi M, Keshavarz M, Kadkhodaee M, Mirershadi F, Asadi B. Anti-infarct effect of magnesium is not mediated by adenosine A1 receptors in rat globally ischaemic isolated hearts. Clin Exp Pharmacol Physiol 2004;31:868-872.
http://dx.doi.org/10.1111/j.1440-1681.2004.04128.x
Monahan TS, Sawmiller DR, Fenton RA, Dobson JGJ. Adenosine A2a-receptor activation increases contractility in isolated perfused hearts. Am J Physiol Heart Circ Physiol 2000;279:H1472–1481.
Park SS, Zhao H, Jang Y, Mueller RA, Xu Z. N6-(3-iodobenzyl)-adenosine-5'-N-methylcarboxamide confers cardioprotection at reperfusion by inhibiting mitochondrial permeability transition pore opening via glycogen synthase kinase 3 beta. J Pharmacol Exp Ther 2006;318(1):124-131.
http://dx.doi.org/10.1124/jpet.106.101477
Kaplan GB L-MK, Sears MT. Alterations of adenosine A1 receptors in morphine dependence. Brain Res 1994;657:347-350.
http://dx.doi.org/10.1016/0006-8993(94)90990-3
Sandner-Kiesling A, Li X, Eisenach JC. Morphine-induced spinal release of adenosine is reduced in neuropathic rats. Anesthesiology 2001;95:1455-1459.
http://dx.doi.org/10.1097/00000542-200112000-00026
Headrick JP, Hack B, Ashton KJ. Acute adenosinergic cardioprotection in ischemic-reperfused hearts. Am J Physiol Heart Circ Physiol 2003;285:H1797–1818.
http://dx.doi.org/10.1152/ajpheart.00407.2003
Kuno A, Critz SD, Cui L, Solodushko V, Yang XM, Krahn T, et al. Protein kinase C protects preconditioned rabbit hearts by increasing sensitivity of adenosine A2b-dependent signaling during early reperfusion. J Mol Cell Cardiol 2007;43(3):262-271.
http://dx.doi.org/10.1016/j.yjmcc.2007.05.016
Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res 2006;70(2):308-314.
Downloads
Additional Files
Published
Issue
Section
Categories
How to Cite
Accepted 2015-10-23
Published 2016-01-01





