ALKBH5 modulates m6A modification to enhance acute myeloid leukemia resistance to adriamycin
DOI:
https://doi.org/10.17305/bb.2024.11076Keywords:
ALKBH5, TUG1, SH3BGRL, EHMT2, acute myeloid leukemia, N6-methyladenosine, H3K9me2Abstract
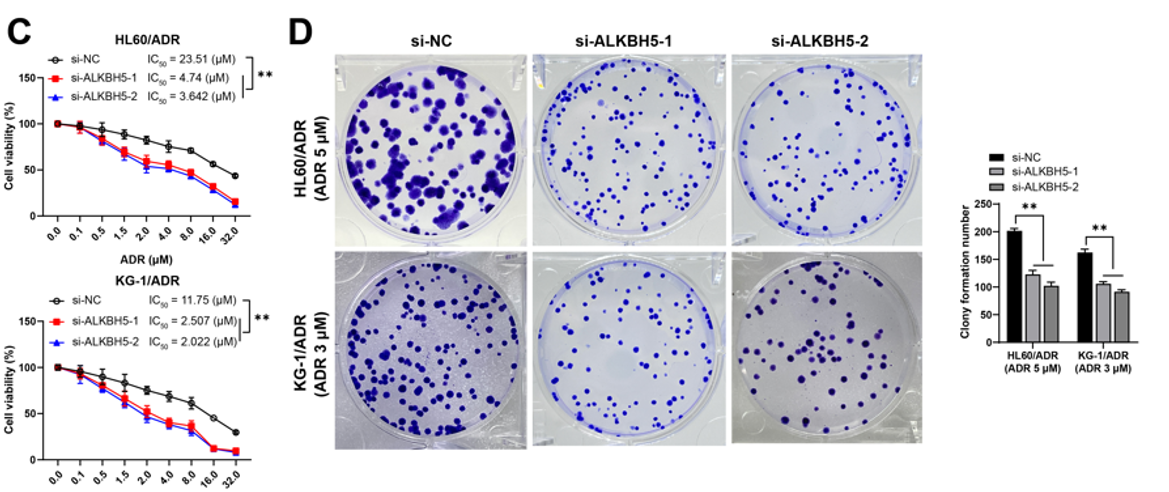
Acute myeloid leukemia (AML) is a fatal malignancy with rising incidence and low cure rates. This study aims to investigate the effect of alkB homolog 5 (ALKBH5)-mediated N6-methyladenosine (m6A) modification on adriamycin (ADR) resistance in AML. First, the levels of ALKBH5, taurine upregulated 1 (TUG1), YTH N6-methyladenosine RNA binding protein F2 (YTHDF2), euchromatic histone lysine methyltransferase 2 (EHMT2), and SH3 domain-binding glutamate-rich protein-like (SH3BGRL) were measured. IC50 values, cell proliferation, and apoptosis were determined. m6A levels were analyzed, and the binding interactions between TUG1 and YTHDF2, as well as TUG1 and EHMT2, were assessed. The stability of TUG1 and the enrichment of EHMT2 and H3K9me2 on the SH3BGRL promoter were confirmed. In vivo experiments were conducted to further validate the results. The findings revealed that ALKBH5 was overexpressed in both AML- and ADR-resistant cells, and silencing ALKBH5 reduced the ADR resistance of AML cells. ALKBH5 removed m6A modifications from TUG1, disrupting the interaction between YTHDF2 and TUG1, thereby stabilizing TUG1 expression. TUG1 bound to EHMT2, promoting H3K9me2 modification on the SH3BGRL promoter and suppressing SH3BGRL expression. Overexpression of TUG1 or knockdown of SH3BGRL reversed the suppressive effect of ALKBH5 knockdown on ADR resistance. In vivo, ALKBH5 knockdown inhibited ADR resistance in AML cells. In conclusion, ALKBH5 removed m6A modification to stabilize TUG1 expression in a YTHDF2-dependent manner, enhancing H3K9me2 levels on the SH3BGRL promoter and suppressing SH3BGRL expression, thus promoting ADR resistance in AML cells.
Citations
Downloads
Downloads
Published
Issue
Section
Categories
License
Copyright (c) 2024 Yonghua Liu, Jinhong Jiang, Yuxiao Zeng, Yu Jiang

This work is licensed under a Creative Commons Attribution 4.0 International License.





