Rare liver diseases — Etiology, diagnosis and management: A review
DOI:
https://doi.org/10.17305/bb.2025.13278Keywords:
Primary biliary cholangitis, primary sclerosing cholangitis, autoimmune hepatitis, gene therapy, precision medicineAbstract
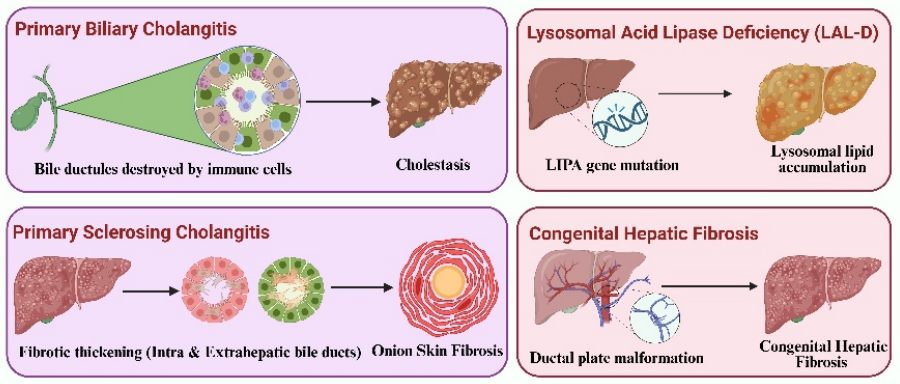
Rare liver diseases (RLDs) are diverse and often misdiagnosed conditions that impose significant clinical and public health challenges due to their variable presentations and limited treatment options. This study aims to synthesize contemporary evidence on the etiology, classification, diagnostics, and management of RLDs and to identify near-term research and implementation priorities. We conducted a systematic search of PubMed and Scopus for the years 2015 to 2025 using predefined keywords. We included peer-reviewed human studies, such as guidelines, randomized trials, and large registries, focusing on mechanisms, diagnostic strategies, and treatments. We excluded animal studies and non-peer-reviewed reports, extracting data on disease biology, diagnostic tools, outcomes, and molecular therapies. RLDs can be categorized into genetic/inherited, autoimmune cholestatic, and other vascular/metabolic entities. Care for these diseases is increasingly guided by structured pathways that integrate biochemistry and serology with magnetic resonance cholangiopancreatography (MRCP), elastography, targeted next-generation sequencing (NGS), and selective biopsy. Emerging biomarkers, such as circulating microRNAs, alongside machine learning in imaging techniques, enhance disease staging and prognostication. Key management strategies include the use of bile-acid modulators, surgical interventions, and ileal bile acid transporter (IBAT) inhibitors for progressive familial intrahepatic cholestasis (PFIC). Lifelong copper chelation is recommended for Wilson disease, with trientine preferred for neurologic phenotypes. Supportive care in alpha-1 antitrypsin deficiency (A1ATD) is complemented by the investigation of molecular chaperones. Additionally, gene-directed therapies, gene editing, RNA-based approaches, and cell therapies show early promise but raise concerns regarding durability, safety, and ethical considerations, particularly for pediatric patients. In conclusion, implementing precision medicine frameworks that rely on standardized diagnostics, multicenter registries, and equitable access is crucial for facilitating earlier detection and translating mechanism-targeted therapies into sustainable, globally accessible benefits.
Citations
Downloads
Downloads
Published
License
Copyright (c) 2025 Qi Long, Rawaz D. Tawfeeq, Yu Jiang, Huabao Liu, Meiao Tan

This work is licensed under a Creative Commons Attribution 4.0 International License.





